Dans la continuité de notre article sur les antifongigrammes, nous poursuivons notre exploration de la mycologie avec un éclairage sur les antifongiques à usage systémique : les classes, leurs cibles, leurs mécanismes d’action et les spécificités d’utilisation. Ces éléments sont essentiels pour interpréter les résultats d’un antifongigramme et adapter au mieux la prise en charge thérapeutique.
Introduction
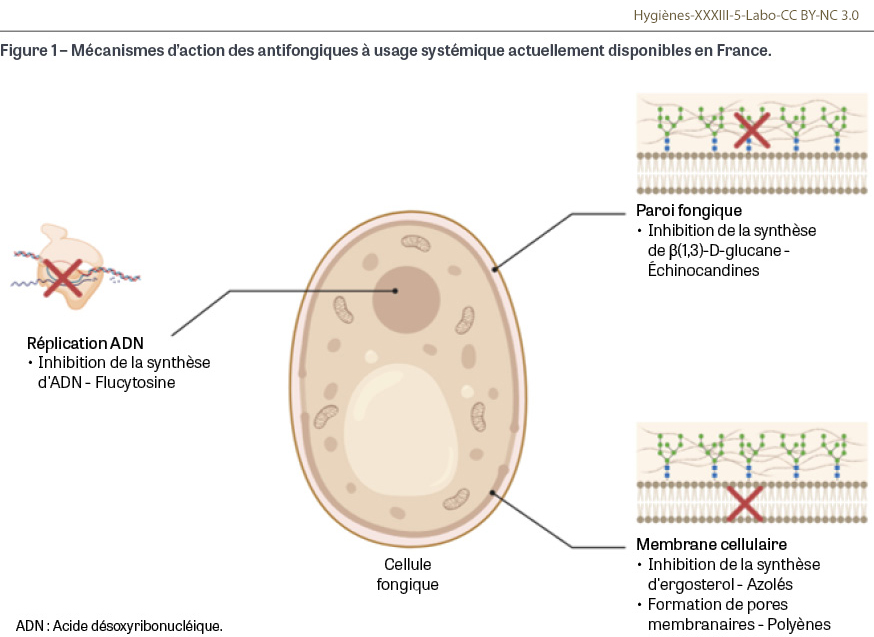
Les infections fongiques invasives (IFI) représentent une menace croissante dans les services de soins hospitaliers, notamment pour les patients immunodéprimés, qu’il s’agisse classiquement de patients d’hématologie ou de patients traités par de nouvelles molécules immunosuppressives, ou hospitalisés en unité de soins intensifs ou encore porteurs de matériel. En 2023, 3 666 cas d’IFI ont été notifiés en France (60 centres participants), dont une grande majorité de fongémies (54%) et une mortalité globale de 39,8% à trois mois pour l’ensemble des IFI [1]. Face à cette menace, le rôle des antifongiques est crucial, mais leur utilisation raisonnée repose avant tout sur une bonne compréhension de leur mécanisme d’action et de leur spectre d’activité. Les polyènes, les antimétabolites, les azolés et les échinocandines représentent les principales classes d’antifongiques actuellement commercialisées. Ces classes se différencient par leurs effets pharmacologiques. Certaines molécules présentent un effet fongicide, qui provoque la mort des cellules fongiques, tandis que d’autres exercent une action fongistatique, qui inhibe la croissance des champignons. Les antifongiques diffèrent également par leur mécanisme d’action, qui leur confère un spectre d’action spécifique (Figure 1). Ainsi, certaines espèces possèdent des résistances naturelles, également nommées intrinsèques, à quelques antifongiques. L’acquisition de résistances extrinsèques par les espèces d’intérêt médical représente un enjeu majeur de santé publique. Aspergillus fumigatus en est un exemple concret, avec des descriptions de résistances aux molécules azolées de plus en plus nombreuses depuis une dizaine d’années [2]. Le nombre de souches de Candida parapsilosis résistantes au fluconazole est également en constante augmentation depuis quelques années, atteignant jusqu’à 27% des isolats impliqués dans les IFI en France, en 2022 [1]. Autre exemple, Candidozyma (ex-Candida) auris, levure émergente, présente des concentrations minimales inhibitrices vis-à-vis des azolés et des échinocandines avec une variabilité importante, certaines pouvant atteindre des valeurs élevées [3].
Polyènes
L’amphotéricine B, unique molécule utilisée par voie systémique au sein de cette classe, constitue un traitement antifongique incontournable dans la lutte contre les IFI. Découverte dans les années 1950, elle s’est rapidement imposée comme une référence, jusqu’à figurer aujourd’hui sur la liste des médicaments jugés essentiels par l’Organisation mondiale de la santé [4]. C’est une molécule complexe et volumineuse qui exerce une action fongicide en interagissant avec l’ergostérol, un composant fondamental de la membrane plasmique de la cellule fongique (Figures 1 et 2). Grâce à sa structure amphiphile, c’est-à-dire à la fois lipophile et hydrophile, l’amphotéricine B est capable de se lier à l’ergostérol (structure proche du cholestérol) de la membrane et de s’y incorporer pour former un pore transmembranaire. Ainsi, un passage se crée entre le cytoplasme et l’environnement extérieur, par lequel s’opère une fuite ionique massive, notamment en potassium. Un déséquilibre des potentiels électriques entre l’intérieur et l’extérieur de la cellule se forme, déstabilisant l’ensemble des mécanismes de survie de la cellule et conduisant à sa mort. L’amphotéricine B désoxycholate (Fungizone®, Bristol Myers-Squibb, New York, NY, États-Unis) est la première forme galénique de la molécule qui a été commercialisée. D’un point de vue purement structurel, il s’agit de l’amphotéricine B associée à un sel biliaire, le désoxycholate de sodium. Celui-ci forme un complexe micellaire afin d’assurer une solubilisation de l’amphotéricine B pour une administration intraveineuse. Bien qu’efficace, cette formulation présente des difficultés d’administration et une toxicité rénale majeure, notamment lorsqu’elle est administrée à forte dose. Cette toxicité dépendante de la dose provoque des atteintes des glomérules et des tubules rénaux qui peuvent être irréversibles. D’autres effets indésirables, également fréquents, peuvent être directement imputés à l’injection du médicament, tels que des nausées, des vomissements, une fièvre, ou encore des manifestations allergiques sévères. Plus rarement, des troubles hépatiques ou une anémie peuvent survenir. Afin de limiter cette toxicité, une nouvelle formulation pharmacologique a été développée : l’amphotéricine B liposomale (AmBisome®, NeXstar Pharmaceuticals, Boulder, CO, États-Unis). Elle associe la molécule active avec des vésicules liposomales (mélange de phospholipides et de cholestérol). L’administration intraveineuse est mieux tolérée, la distribution tissulaire est meilleure et la demi-vie, plus longue. Elle est aujourd’hui bien plus souvent prescrite que son homologue, malgré un coût plus élevé. L’amphotéricine B liposomale est active contre de nombreux champignons filamenteux, ce qui en fait une alternative de seconde intention en cas d’aspergillose ou de fusariose invasive [5,6]. Elle est également utilisée en traitement de première intention, en association avec une prise en charge chirurgicale, dans les infections à Mucorales [7]. Elle trouve aussi une application dans le traitement des infections fongiques sévères à levures grâce à son activité fongicide. Sa diffusion dans le liquide cérébro-spinal est limitée du fait de sa taille et de sa nature, mais elle reste un élément clé dans le traitement de la cryptococcose neuro-méningée. Néanmoins, son spectre ne couvre pas toutes les espèces fongiques. Certaines d’entre elles y sont naturellement résistantes, qu’il s’agisse de levures (Trichosporon spp.) ou de champignons filamenteux (Scedosporium spp., Aspergillus terreus). La résistance à l’amphotéricine B reste rare et mal explorée, même si plusieurs mécanismes ont été évoqués, dont le système de réponse au stress oxydatif ou au stress thermique [8]. Bien qu’exceptionnelles, certaines résistances acquises ont même été décrites après exposition, comme chez certaines souches de Clavispora lusitaniae (ex-Candida lusitaniae), par une mutation du gène ERG6, codant des protéines impliquées dans la synthèse de l’ergostérol [9].
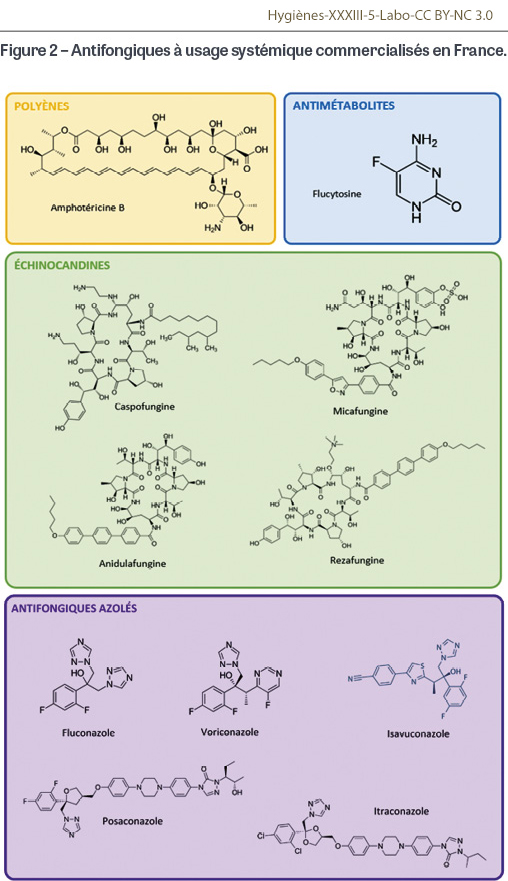
Antimétabolites
La 5-fluorocytosine (5-FC ; Ancotil®, Viatris, Canonsburg, PA, États-Unis), ou flucytosine, est une prodrogue qui se transforme en un métabolite possédant l’activité antifongique. Administrée par voie orale, la 5-FC est captée par la cellule fongique où elle est métabolisée en 5-fluoro-uracile (5-FU). Celui-ci, analogue de base pyrimidique, est capable de s’incorporer dans l’ADN1 lors de sa réplication, grâce à l’action de l’ADN polymérase, pour ensuite bloquer le cycle cellulaire. Le 5-FU est efficace contre l’ensemble des cellules eucaryotes du monde vivant, y compris les cellules humaines, et est parfois utilisé dans des protocoles thérapeutiques anticancéreux pour son activité antimitotique. De ce fait, il est important de noter que la 5-FC n’est métabolisée en 5-FU que par les cellules fongiques, ce qui en limite théoriquement les effets sur les cellules humaines. La flucytosine peut néanmoins être à l’origine de certains effets indésirables et entraîner des altérations cardiaques, habituellement d’allure ischémique, ou neurologiques de type céphalées, sédations ou convulsions. Du fait de sa fonction antimétabolique, la flucytosine peut également être à l’origine de troubles hématopoïétiques, tels que thrombopénie, anémie ou leucopénie, imposant une surveillance stricte. Ce médicament est par ailleurs contre-indiqué chez la femme enceinte, en raison d’un risque de malformation fœtale. La flucytosine possède une action fongistatique sur les levures. Elle doit toujours être utilisée en association avec un autre antifongique car elle est sujette à un fort risque d’émergence de souches résistantes. Elle est administrée en association avec l’amphotéricine B dans le traitement d’attaque de première intention de la cryptococcose neuro-méningée [10]. La flucytosine peut aussi être utile en association dans la prise en charge d’infections disséminées à Candida, telles que des endocardites ou des infections du système nerveux central [11]. Son spectre d’activité reste tout de même limité en raison de nombreuses résistances intrinsèques et potentielles, comme chez Trichosporon spp. ou Clavispora lusitaniae. Au final, la flucytosine fait partie des options thérapeutiques les moins utilisées.
Azolés
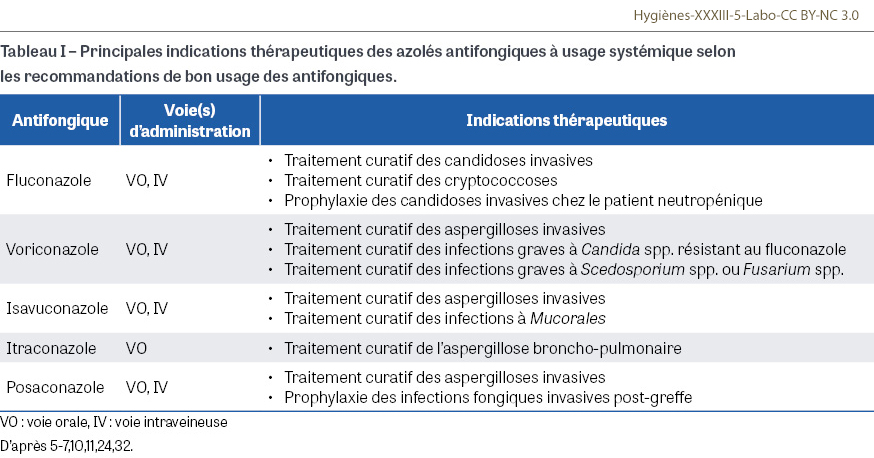
Les antifongiques azolés constituent une classe médicamenteuse hétérogène, tant par la diversité des molécules disponibles que par leurs caractéristiques pharmacologiques. Les azolés à usage systémique (fluconazole, voriconazole, isavuconazole, itraconazole, posaconazole) partagent un même mécanisme d’action : l’inhibition de la synthèse de l’ergostérol, composant essentiel de la membrane fongique, par blocage sélectif de la 14-α-déméthylase codée par les gènes CYP51 chez les champignons filamenteux ou ERG chez les levures. Cette inhibition confère à l’ensemble de la classe une action fongicide sur la plupart des champignons filamenteux — à l’exception notable du fluconazole, inefficace sur ces derniers et qui n’a qu’une action fongistatique sur les levures. Les azolés à usage systémique sont utilisés en traitement curatif mais également en prophylaxie chez les patients à haut risque, notamment chez les patients d’hématologie (Tableau I). Sur le plan des effets indésirables, les azolés partagent un profil commun. L’hépatotoxicité est fréquente, se manifestant par une cytolyse ou une cholestase. Des troubles digestifs sont également régulièrement rapportés. Plus rarement, ces médicaments peuvent entraîner un allongement de l’intervalle QT à l’électrocardiogramme, exposant à un risque de torsade de pointes. Par ailleurs, le fluconazole, le voriconazole et l’isavuconazole peuvent provoquer des effets neurologiques, dont la nature et la fréquence varient selon la molécule. Enfin, il est important d’évoquer la toxicité cutanée du voriconazole, allant du rash jusqu’au carcinome épidermoïde, effet indésirable notoire pouvant conduire à l’arrêt du traitement [12]. L’efficacité du traitement peut être influencée par de nombreux facteurs individuels. Outre la sensibilité intrinsèque du champignon, l’activité variable des cytochromes peut modifier les concentrations plasmatiques et la réponse au traitement. Le voriconazole en est un bon exemple, car il est sujet à une forte variabilité interindividuelle, provoquée par un polymorphisme génétique des cytochromes CYP2C19 et CYP3A4, pouvant induire son hypermétabolisation [13]. Il est également sensible à des facteurs ponctuels (alimentation, diarrhées…) ou à des états physiopathologiques particuliers. Par exemple, la concentration plasmatique du voriconazole est corrélée au taux sanguin de protéine C-réactive (CRP) : l’inflammation induit une diminution du métabolisme de la molécule, donc une concentration accrue, par le biais du CYP450, moins actif [14]. Des résistances acquises aux azolés peuvent également émerger à la suite d’une exposition prolongée en contexte médical, ou d’un usage intensif de fongicides en agriculture. Ces pressions sélectives favorisent l’apparition de mutations, notamment dans le gène CYP51A qui code l’enzyme cible des azolés [15]. La résistance d’Aspergillus fumigatus aux azolés, de plus en plus fréquemment observée, représente un exemple préoccupant du concept One Health (une seule santé). En effet, l’usage agricole de fongicides azolés exerce une pression sélective sur les champignons environnementaux, dont Aspergillus fumigatus. Cet usage favorise la sélection de souches résistantes susceptibles d’être inhalées par les patients qui ne pourront alors plus être traités par les azolés, pourtant considérés comme le traitement de première ligne des aspergilloses invasives [6,16,17]. Chez Candida albicans, la survenue de nouvelles substitutions dans la séquence génétique du gène ERG11, codant la 14-α-déméthylase, provoque une diminution de la sensibilité aux azolés antifongiques [18,19,20]. Chez d’autres espèces de levures (Candida tropicalis, Candida parapsilosis, Pichia kudriavzevii…), d’autres mutations identifiées sur les gènes intervenant dans la synthèse d’ergostérol ont également montré une augmentation des concentrations minimales inhibitrices pour plusieurs antifongiques azolés [20]. Un suivi des concentrations plasmatiques résiduelles est recommandé pour le posaconazole, l’itraconazole, l’isavuconazole et le voriconazole, compte tenu des caractéristiques précédemment citées et de la corrélation entre concentration plasmatique et efficacité clinique. Ce suivi thérapeutique doit être complété par une évaluation régulière des réponses clinique, biologique et radiologique, ainsi que par une surveillance rigoureuse des effets indésirables, notamment par la réalisation d’un bilan hépatique [21].
Échinocandines
Isolée pour la première fois en 1974 à partir d’une souche d’Aspergillus, l’échinocandine B a été progressivement modifiée pour donner naissance à une classe thérapeutique aujourd’hui incontournable dans le traitement des IFI. Actuellement, quatre molécules destinées à la voie intraveineuse sont commercialisées en France : la caspofungine (Cancidas®, Merck, Darmstadt, Allemagne), la micafungine (Mycamine®, Sandoz, Bâle, Suisse), l’anidulafungine (Ecalta®, Pfizer, New York, NY, États-Unis) et la rézafungine (Rezzayo®, Mundipharma, Cambridge, Royaume-Uni). Cette dernière, disponible depuis 2024 en France, possède une action prolongée, ce qui permet des injections hebdomadaires au lieu des injections quotidiennes des autres échinocandines [22]. Ces antifongiques se distinguent des autres par un mode d’action unique, empêchant la synthèse du (1,3)-β-D-glucane, un composant majeur de la paroi fongique, en inhibant l’enzyme (1,3)-β-D-glucane synthase dont les sous-unités sont codées par trois gènes FKS. En déstabilisant cette paroi, les échinocandines exercent une action fongicide contre la majorité des levures, une action fongistatique sur certains champignons filamenteux sensibles, ainsi qu’une efficacité notable contre certains biofilms, notamment ceux de Candida albicans et Nakaseomyces glabratus (ex-Candida glabrata) [23]. En revanche, elles sont inefficaces contre les champignons dépourvus de (1,3)-β-D-glucane, comme c’est le cas pour les espèces des genres Cryptococcus et Trichosporon, ou encore pour les Mucorales. De plus, certaines espèces filamenteuses, comme Lomentospora prolificans, sont résistantes par des mutations génétiques dans les régions hautement conservées (hot-spots) HS1 et HS2 du gène FKS1, cible des échinocandines. Administrées par voie intraveineuse en perfusion, les échinocandines sont indiquées en première intention dans le traitement curatif des candidoses invasives. Outre leur usage curatif, elles peuvent être employées de manière empirique chez les patients neutropéniques à haut risque d’IFI [24]. C’est d’ailleurs une des raisons pour lesquelles ce sont des molécules fréquemment prescrites dans les services d’hématologie et de réanimation. Le suivi des patients traités par échinocandines est important. L’amélioration clinique et radiologique témoigne de l’efficacité du traitement, mais également de la tolérance à celui-ci. Parallèlement, une surveillance biologique est indispensable, incluant les paramètres de l’ionogramme, les marqueurs hépatiques (transaminases, phosphatases alcalines, gamma-GT2) et la formule sanguine. Ce suivi est nécessaire car les échinocandines peuvent être à l’origine d’effets indésirables importants, tels que des troubles hépatiques, une anémie ou des désordres hémodynamiques, notamment en cas de surdosage. Des céphalées et des troubles digestifs sont souvent rapportés, tandis que des éruptions cutanées et, plus rarement, des convulsions, peuvent survenir. Un suivi pharmacologique est recommandé uniquement dans certaines situations particulières pour s’assurer d’une concentration sanguine optimale du médicament [25]. Par ailleurs, l’utilisation de certains dispositifs médicaux peut être à l’origine d’une séquestration importante de la caspofungine et rendre le traitement inefficace. Par exemple, depuis 2024, par suite de l’analyse de plusieurs signalements, l’Agence nationale de sécurité du médicament (ANSM) recommande de ne pas utiliser de membranes d’épuration extra-rénale constituées de dérivés du polyacrylonitrile chez les patients en soins intensifs [26].
Nouveaux antifongiques
Malgré l’efficacité des drogues antifongiques actuellement disponibles, leur spectre parfois limité, la toxicité de certains agents, le coût élevé des traitements journaliers par échinocandine et par amphotéricine B liposomale, ainsi que l’émergence de résistances fongiques imposent le développement de nouvelles molécules. Grâce à des recherches de thérapies innovantes ciblant des mécanismes d’action inédits ou optimisant la pharmacocinétique et la tolérance des traitements existants, plusieurs candidats sont aujourd’hui en phase avancée de développement préclinique ou clinique.
Olorofim
L’Olorofim (F2G, Macclesfield, Royaume-Uni) fait partie d’une toute nouvelle classe thérapeutique : les orotomides. C’est un inhibiteur sélectif de la dihydro-orotate déshydrogénase (DHODH) fongique, une enzyme impliquée dans la synthèse des bases pyrimidiques nécessaires à la cellule fongique pour la synthèse d’ADN et d’ARN3 [27]. Non commercialisé à ce jour, l’Olorofim est une molécule en fin de phase II de développement. Une étude menée en 2018 a montré que l’Olorofim permet une inhibition précoce de la germination de la cellule fongique, l’empêchant de former des filaments donc de se développer [28]. Une exposition prolongée des cellules fongiques a également révélé une activité fongicide sur les champignons filamenteux et une bonne diffusion tissulaire, notamment cérébrale. Plus tard, des essais sur certaines espèces hautement résistantes aux antifongiques usuels (Lomentospora prolificans, Scedosporium sp., Exophiala dermatitidis, Rasamsonia sp. et Aspergillus fumigatus résistants aux azolés) ont montré une bonne efficacité antifongique. Les essais sur les levures et les Mucorales ont été rapidement abandonnés car elles semblent insensibles à l’Olorofim [29].
Ibrexafungerp
L’ibrexafungerp est un nouvel inhibiteur de la (1,3)-β-D-glucane synthase de la classe des triterpénoïdes. Certains premiers résultats de phase II démontrent une efficacité thérapeutique et une bonne tolérance dans le traitement des candidoses invasives, en comparaison d’un traitement par échinocandine [30]. Des essais cliniques de phase III sont en cours de finalisation. Avec des interactions médicamenteuses minimes et une administration par voie orale ou intraveineuse, l’ibrexafungerp semble se dessiner comme candidat crédible en relais dans le traitement des candidoses invasives ou dans le traitement des aspergilloses invasives.
Fosmanogepix
Le Fosmanogepix (Basilea Pharmaceutica, Bâle, Suisse) est une prodrogue dont le métabolite est un inhibiteur de la GWt1, enzyme acétyltransférase impliquée dans la synthèse des mannoprotéines membranaires contribuant à la stabilité de la membrane plasmique de la cellule fongique. Son spectre d’activité est large, couvrant des levures (Cryptococcus spp., Candida spp. et Candidozyma auris) et des champignons filamenteux (Aspergillus résistant aux azolés, Fusarium spp., Scedosporium spp., Lomentospora prolificans). De plus, le Fosmanogepix montre des résultats encourageants concernant sa diffusion dans le liquide cérébro-spinal [31]. Une étude clinique de phase III est en cours dans le traitement des candidémies et des candidoses invasives. Il est actuellement disponible en autorisation d’accès compassionnel auprès de l’ANSM sous forme orale ou intraveineuse.
Conclusion
Les antifongiques actuels restent essentiels mais présentent souvent des limites, notamment en termes de toxicité et d’émergence de résistances. Les nouvelles molécules offrent des modes d’action inédits qu’il conviendra d’analyser, notamment sous l’angle des résistances. L’avenir du traitement des infections fongiques repose donc sur une optimisation de leur efficacité, de leur tolérance et de la prévention des résistances.
Notes :
1- Acide désoxyribonucléique.
2- Gamma-glutamyltransférase.
3- Acide ribonucléique.