Introduction
Nous poursuivons et concluons dans ce nouveau numéro la description des différentes classes d’antibiotique, en incluant les grandes indications et l’incidence de la résistance à ces molécules chez les principales bactéries d’intérêt médical. Dans le précédent numéro nous nous étions intéressés aux bêtalactamines, pilier des antibiothérapies empiriques ou basées sur les résultats de l’antibiogramme. La résistance naturelle ou acquise aux pénicillines et aux céphalosporines ou la nécessité de les administrer pour les molécules à plus large spectre par voie parentérale ont pour conséquence le besoin de disposer d’alternatives à cette classe majeure d’antibiotique. De même que nous l’avions déploré pour les bêtalactamines, ces « autres » antibiotiques sont assez peu innovants : la description de la dernière nouvelle classe représentée par les oxazolidinones et le linézolide remonte en effet aux années 2000. Des molécules anciennes, mises de côté car trop toxiques comme la colistine ou la daptomycine, ont dû être réhabilitées pour faire face à l’apparition de nouvelles bactéries multirésistantes. La particularité des antibiotiques, seule classe de médicaments à voir son efficacité diminuer avec le temps, en fait le parent pauvre des programmes de recherche des sociétés pharmaceutiques. À l’instar des bêtalactamines, les autres molécules antibiotiques ont pour la plupart une origine naturelle, qu’elles soient produites par des bactéries ou par des champignons (ce qui justifie alors le suffixe « mycine ») ; elles ont été ensuite modifiées chimiquement pour optimiser leur efficacité ou leur biodisponibilité ou encore améliorer leur tolérance. Certains composés proviennent néanmoins de l’ingénierie chimique, comme les sulfamides et les fluoroquinolones. Tous ces antibiotiques alternatifs sont concernés par l’apparition et la diffusion de résistances acquises (Tableau I). Leur utilisation seule ou en association doit donc être aussi parcimonieuse et optimisée que celle des bêtalactamines, avec des schémas thérapeutiques les plus courts possibles et à des doses limitant la sélection de mutants résistants.
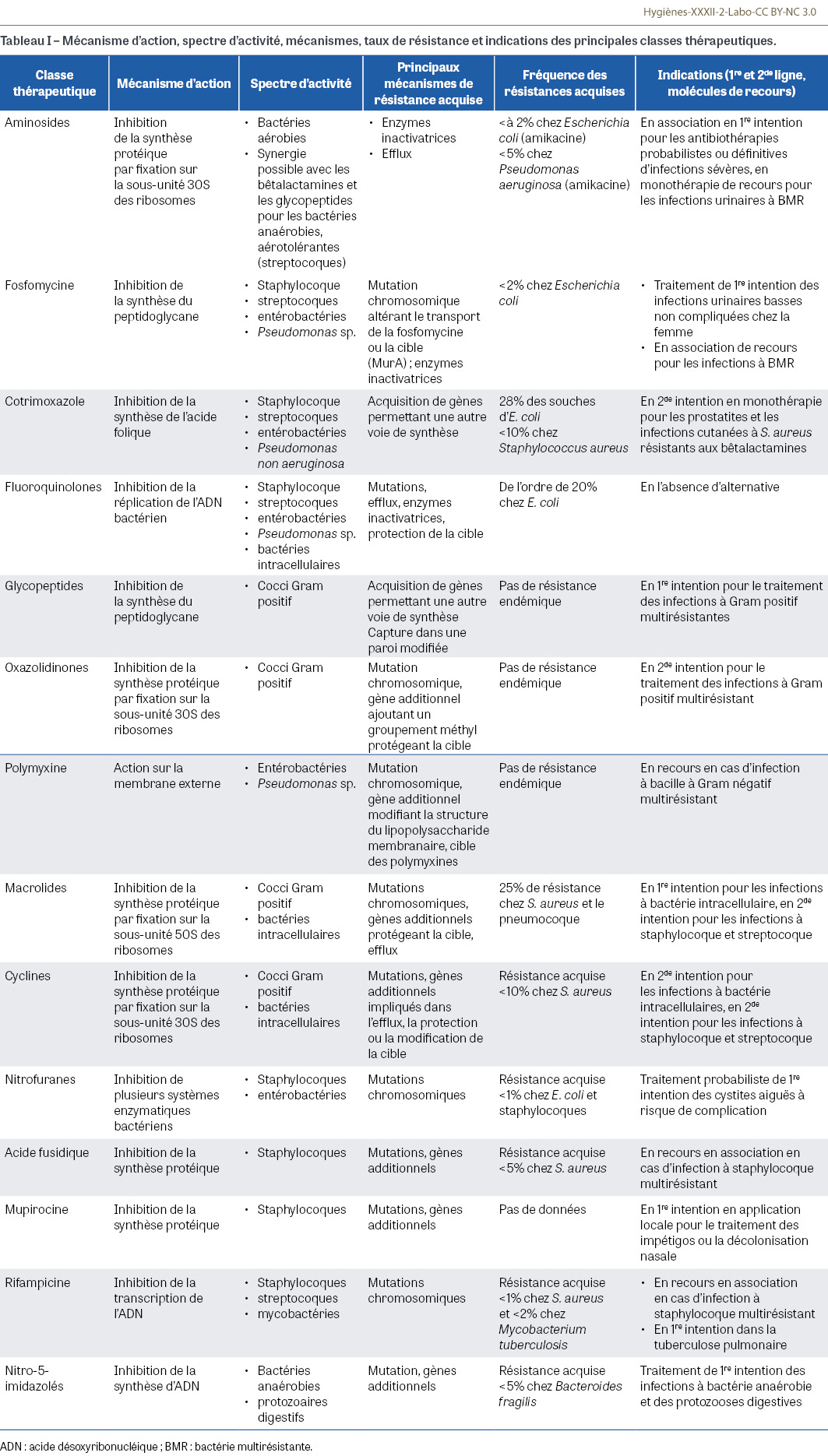
Les aminosides
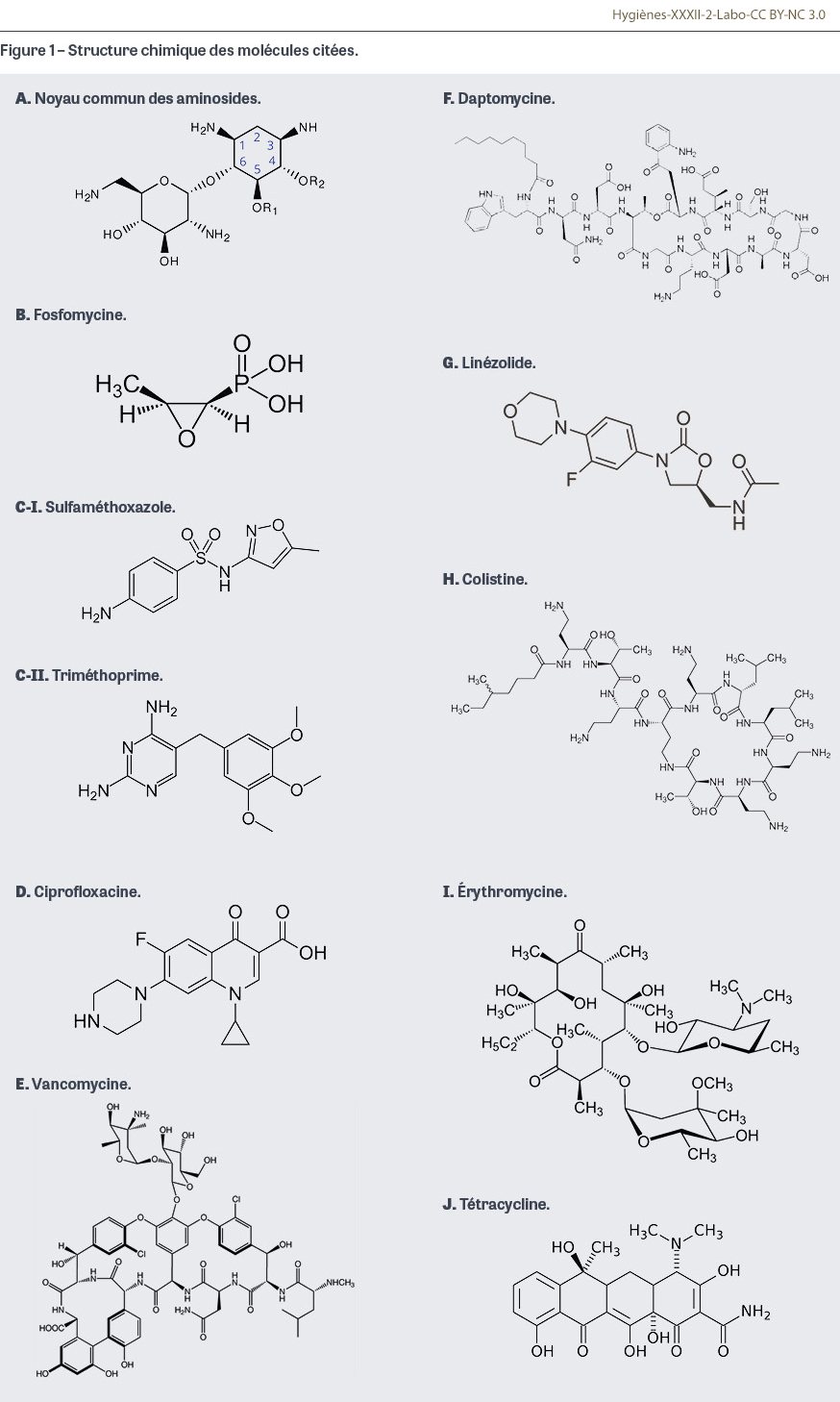
Les aminosides, également appelés aminoglycosides, sont des antibiotiques développés pendant la Seconde Guerre mondiale. La streptomycine a été découverte en 1943 par Selman Abraham Waksman qui reçut pour cela le prix Nobel en 1952. Ces molécules sont composées de sucres (« osides ») substitués par des fonctions aminées (-NH2), d’où leur appellation. Le noyau central est composé de 2-désoxystreptamine et de glucosamine qui sont ensuite substituées par d’autres sucres aminés en position 4 ou 5 (Figure 1A). Ce sont des molécules naturelles produites par des bactéries, les actinomycètes et les streptomycètes. Les molécules naturelles présentent une ototoxicité et une toxicité rénale (néomycine, kanamycine…). Aujourd’hui ces molécules ne sont plus utilisées qu’en application locale (pommade, gouttes auriculaires si le tympan n’est pas percé, ou sous la forme de collyre), ou par voie orale afin de décontaminer l’intérieur du tube digestif car elles ne sont pas absorbées. Des dérivés modifiés chimiquement ont été développés et font toujours partie des stratégies thérapeutiques actuelles : gentamicine, amikacine, netilmicine, tobramycine. Le mode d’action principal des aminosides est l’inhibition de la première étape de la synthèse protéique (l’initiation) par fixation sur la sous-unité 30S des ribosomes bactériens. Les molécules pénètrent dans la bactérie en traversant la membrane cytoplasmique par un mécanisme oxygéno-dépendant : les bactéries anaérobies strictes, c’est-à-dire qui ne se développent qu’en l’absence d’oxygène, sont donc naturellement résistantes. Les entérocoques et les streptocoques, bactéries anaérobies qui tolèrent l’oxygène, sont également naturellement résistants ; néanmoins en présence de bêtalactamines, les aminosides retrouvent une activité dite « synergique ». L’association bêtalactamine-aminoside constitue donc un cocktail très efficace lorsqu’une action rapide et fortement bactéricide est recherchée, notamment lors d’infections graves comme les endocardites. Cette association est également utilisée en antibiothérapie empirique ou probabiliste ciblant une infection à bacille à coloration de Gram négative (entérobactérie, Pseudomonas aeruginosa) lorsqu’une résistance acquise à la bêtalactamine est possible (par production d’une bêtalactamase à spectre étendu chez une entérobactérie par exemple). Cette association est le plus souvent limitée à quelques jours, le temps d’obtenir l’antibiogramme ou de diminuer l’inoculum bactérien : les aminosides restent des molécules toxiques pour le rein et l’oreille interne. Au-delà de quelques jours, il convient de les doser dans le sang afin de s’assurer de leur efficacité (prélever au pic sérique, soit 30 minutes après l’injection) et de leur toxicité (valeur résiduelle prélevée juste avant une nouvelle injection) [1]. Les aminosides sont des antibiotiques dont l’activité est « concentration-dépendante », c’est-à-dire associée à la concentration maximale de la molécule dans le sang. Ils sont donc administrés en une seule injection rapide, et jamais en perfusion continue. Il est à noter que des administrations par inhalation ont été développées en prévention ou en traitement d’infections respiratoires chez des patients atteints de mucoviscidose (tobramycine) ou intubés et ventilés en réanimation (amikacine). Les aminosides sont efficaces en théorie contre les staphylocoques, les Listeria, les entérobactéries, P. aeruginosa… En plus des bactéries anaérobies, des bactéries de l’environnement comme Stenotrophomonas maltophilia et Burkholderia cepacia sont également naturellement résistantes. Les résistances acquises se sont développées via des enzymes inactivant les aminosides. La gentamicine reste le plus souvent efficace sur les bactéries à Gram positif et l’amikacine contre celles à Gram négatif : en fonction des espèces ciblées, l’une ou l’autre de ces molécules sera choisie préférentiellement lors des antibiothérapies probabilistes. Néanmoins des enzymes inactivant la totalité des molécules sont apparues et ont diffusé au sein notamment d’entérobactéries multirésistantes. Chez P. aeruginosa, l’amikacine et la tobramycine sont des molécules naturellement actives mais susceptibles d’être inactivées par des pompes d’efflux. En 2022, le pourcentage de souches isolées d’hémocultures avec une résistance acquise à l’amikacine était de 1,5% pour Escherichia coli, 4,1% pour Klebsiella pneumoniae et 4,8% pour E. cloacae complex [2]. Chez Pseudomonas aeruginosa, ce taux de résistance acquise atteignait 4,0% en 2022 [2]. Comme évoqué précédemment, les aminosides sont très majoritairement administrés en association avec une bêtalactamine ou un autre inhibiteur de la paroi (peptidoglycane) des bactéries comme la vancomycine. Néanmoins, dans certaines situations particulières (infection urinaire basse avec une entérobactérie multirésistante), ils peuvent être utilisés en monothérapie [3]. Leur place restant majeure dans l’arsenal thérapeutique curatif antibactérien, les utilisations en prophylaxie ou en traitement local tendent à diminuer. Toutefois, dans le cadre de l’antibioprophylaxie avant une chirurgie colorectale, les recommandations sont désormais d’administrer la veille de l’intervention 200 mg de tobramycine en complément du métronidazole [4]. Deux molécules méritent d’être citées à titre historique : la streptomycine qui a été à la base du traitement de la tuberculose, avec les inconvénients liés à l’utilisation d’une molécule ototoxique et néphrotoxique pour des périodes longues comme cela est nécessaire au cours de cette pathologie ; et la spectinomycine qui constituait une solution intéressante pour le traitement en une seule injection (traitement dit minute) des infections à gonocoques multirésistantes. Ces antibiotiques ne sont plus commercialisés en France.
La fosfomycine
La fosfomycine a été découverte dans les années 1970 (Figure 1B). Elle est produite par des bactéries du genre Streptomyces. Elle fait partie des antibiotiques agissant sur la paroi des bactéries, c’est-à-dire le peptidoglycane, au même titre que les bêtalactamines et les glycopeptides. Cet antibiotique cible MurA, l’enzyme qui a pour fonction de catalyser la première étape de l’assemblage du peptidoglycane, et inhibe irréversiblement la biosynthèse des parois cellulaires, conduisant à la mort cellulaire. La fosfomycine a une place significative dans l’arsenal thérapeutique. Sous sa forme orale (fosfomycine trométamol), elle constitue le traitement probabiliste monodose de la cystite aiguë simple sans aucun facteur de risque de complication, y compris chez la femme enceinte [5]. Sa très bonne efficacité contre Escherichia coli, la principale espèce responsable des infections urinaires, et sa forte concentration pendant plusieurs jours dans les urines après une seule dose lui permettent d’être largement utilisée dans cette indication, y compris sur délivrance par le pharmacien en cas de positivité d’un test utilisant une bandelette urinaire. Son activité diminuée chez d’autres agents d’infection urinaire tels que les espèces d’entérobactéries Klebsiella pneumoniae et Morganella morganii et la résistance naturelle de Staphylococcus saprophyticus limitent son utilisation dans d’autres situations cliniques. Sous sa forme injectable, la fosfomycine est utilisée en association dans certaines infections graves difficiles à traiter et mettant en cause des bactéries multirésistantes après vérification de leur sensibilité par un antibiogramme. La quantité importante de sels apportée à chaque perfusion et des difficultés d’approvisionnement de la forme injectable de fosfomycine limitent cette utilisation. Depuis la dernière révision des recommandations pour l’antibioprophylaxie, la fosfomycine est recommandée lors de la réalisation de biopsies de prostate par voie transrectale à la dose unique de 3 g per os au moins deux heures avant le geste, en remplacement des fluoroquinolones [4]. La résistance acquise des souches d’E. coli tous prélèvements confondus en 2022 était de 2% [2].
L’association triméthoprime et sulfaméthoxazole (appelée cotrimoxazole)
Les sulfamides ou sulfonamides antibactériens ont été les premiers antibiotiques entièrement synthétiques utilisés en pathologie humaine dès 1935. Ils agissent en bloquant la synthèse bactérienne de l’acide folique. En bactériologie médicale humaine, les sulfamides sont utilisés quasi exclusivement en association avec le triméthoprime, une diaminopyrimidine qui interfère également dans la synthèse bactérienne de l’acide folique. L’association triméthoprime-sulfaméthoxazole a ainsi bénéficié d’une dénomination commune internationale qui lui est propre, le cotrimoxazole, associant ces deux molécules dans un rapport fixe de 1/5 de triméthoprime pour 4/5 de sulfaméthoxazole (Figure 1C). L’utilisation de cette association a été et reste extrêmement large, notamment dans les pays en voie de développement où elle est utilisée dans de très nombreuses indications. En France, le développement d’autres molécules antibiotiques et l’apparition de rares mais graves réactions allergiques de type toxidermies cutanées (syndrome de Lyell ou de Stevens-Johnson) ont considérablement diminué son utilisation au profit notamment des bêtalactamines. Néanmoins son excellente diffusion dans les urines et dans la prostate en fait une molécule intéressante dans ces indications. Les cas d’infection cutanée à Staphylococcus aureus résistant à la méticilline (SARM) communautaire ont également pu bénéficier de cette association. Le cotrimoxazole est aussi largement prescrit en prophylaxie de maladies opportunistes chez l’immunodéprimé, et notamment la pneumocystose, infection à Pneumocystis jirovecii due à un champignon atypique, cause fréquente et redoutable de pneumonie notamment chez les patients greffés. Cette large utilisation a pour conséquences des taux de résistance qui ne permettent pas une utilisation probabiliste sans s’assurer au préalable de la sensibilité du micro-organisme au cotrimoxazole par la réalisation d’un antibiogramme. Le pourcentage de souches d’E. coli isolées d’hémocultures résistantes au cotrimoxazole était de 28,2% en 2022 ; un taux similaire était rapporté pour K. pneumoniae et E. cloacae complex [2]. Dans le cadre de la politique de désescalade antibiotique promue en France depuis quelques années, le triméthoprime seul est de nouveau commercialisé, prescrit et testé pour traiter des infections urinaires basses.
Les quinolones et les fluoroquinolones
La classe d’antibiotique des quinolones et des fluoroquinolones a connu une évolution avec plusieurs générations, atteignant un apogée pour ses indications dans les années 2000. L’acide nalidixique, composé synthétique mis sur le marché au début des années 1960 pour le traitement des infections urinaires basses, est le principal représentant des quinolones. Son mode d’action est l’inhibition de l’ADN1 gyrase bactérienne impliquée dans l’enroulement de l’ADN lors de sa réplication et de sa transcription. Seules les entérobactéries étaient sensibles à cet antibiotique qui se concentrait dans les urines mais ne passait pas dans la circulation générale et ne permettait donc pas de traiter des infections systémiques. Cet antibiotique était administré par voie orale. Les fluoroquinolones ont été découvertes et commercialisées dans les années 1980. Elles possèdent un double mécanisme d’action associant l’inhibition de l’ADN gyrase à celle de la topoisomérase, enzymes indispensables à la réplication de l’ADN bactérien. Cette nouvelle génération présente l’avantage d’avoir une absorption et une diffusion grandement améliorées permettant le traitement d’infections généralisées potentiellement bactériémiantes : infection urinaire haute, infection digestive, pneumopathie… Parmi ces molécules, la ciprofloxacine (Figure 1D) se différencie de l’ofloxacine par son activité sur Pseudomonas aeruginosa. La norfloxacine est quant à elle mal absorbée : elle a été largement utilisée chez les patients cirrhotiques pour diminuer la prolifération bactérienne intra-intestinale et prévenir les translocations digestives et les infections du liquide d’ascite. Le spectre de cette première génération de fluoroquinolones couvre entre autres les staphylocoques, les bactéries intracellulaires (légionelles, Chlamydia, mycoplasmes…), les entérobactéries et, pour la ciprofloxacine, Pseudomonas aeruginosa et Stenotrophomonas maltophilia. En revanche, elles ont une mauvaise activité sur le pneumocoque et les bactéries anaérobies. À la fin des années 1990 sont apparues des fluoroquinolones dites de deuxième génération à activité antipneumococcique, dont la lévofloxacine et la moxifloxacine. Au cours des quinze dernières années, des effets secondaires graves ont été rapportés avec l’usage intensif de cette famille d’antibiotique : tendinopathies, douleurs musculaires et troubles de la marche, neuropathies périphériques, troubles neuropsychiatriques, troubles cardiovasculaires dont des anévrismes, des dissections aortiques, des régurgitations ou insuffisances des valves cardiaques, ou des troubles du rythme. En 2023, suite à de nombreux signalements de pharmacovigilance, l’Agence nationale de la sécurité du médicament et des produits de santé a recommandé de réserver les fluoroquinolones aux infections sévères pour lesquelles il n’existait pas d’alternative [6]. En parallèle de ces données de pharmacovigilance, des résistances acquises se sont développées, principalement par mutations successives des gènes codant la topoisomérase IV et l’ADN gyrase, mais aussi via des pompes d’efflux. Des mécanismes de résistance transférables ont été également mis en évidence via les gènes de la famille qnr (quinolone résistance) qui provenaient de bactéries aquatiques mais aussi via des enzymes inhibant initialement les aminoglycosides. En 2022-2023, la fréquence de la résistance aux fluoroquinolones variait selon l’origine des souches : pour les souches d’E. coli isolées de prélèvements urinaires chez les patients vivant à domicile, le taux de résistance nationale était de 13,5%, alors que pour celles isolées d’hémocultures il était de 17,7%, et qu’il atteignait 19,2% pour celles isolées des prélèvements urinaires de patients vivant en établissement d’hébergement pour personnes âgées dépendantes [2,7]. Pour faire face à ces résistances acquises, de nouvelles molécules ont été récemment commercialisées comme la délafloxacine. Leur intérêt reste à préciser. Les fluoroquinolones sont également utilisées pour le traitement des infections à mycobactérie atypique et des tuberculoses multirésistantes (moxifloxacine).
Les glycopeptides et les lipopeptides
La commercialisation des pénicillines M (méticilline, oxacilline) pour traiter les infections à S. aureus producteur de pénicillinase en 1959 a entraîné dès l’année suivante l’émergence de souches résistantes à la méticilline (SARM). Le support génétique de cette résistance est médié par la présence du gène mecA codant une nouvelle protéine liant la pénicilline, la plp2a. À ce jour, seuls trois types de gènes mec ont été décrits : mecA, mecB retrouvé chez certaines souches de staphylocoques à coagulase négative résistantes à la méticilline et mecC retrouvé chez des souches de S. aureus d’origine animale. Du côté des entérocoques, la résistance acquise fréquente des Enterococcus faecium à l’ampicilline (plus de 75% des souches hospitalières) a nécessité de trouver d’autres solutions que les bêtalactamines pour lutter contre les bactéries à Gram positif multirésistantes aux antibiotiques [2]. Les glycopeptides sont des molécules de grande taille initialement produites par des bactéries de l’environnement (Amycolatopsis orientalis). Ils agissent sur la formation du peptidoglycane. La vancomycine (Figure 1E) est le principal représentant de cette classe d’antibiotique qui n’est pas absorbée par voie orale. La vancomycine s’administre par voie intraveineuse avec la nécessité d’un accès central en raison de sa toxicité périphérique veineuse. Néanmoins, la vancomycine par voie orale constitue l’un des traitements de référence des colites à Clostridioides difficile. La téicoplanine est un glycopeptide moins néphrotoxique, administrable par voie sous-cutanée et présentant une demi-vie permettant une administration unique quotidienne. Des résistances acquises plus fréquentes lui font souvent préférer la molécule princeps. Plus récemment, des glycopeptides semi-synthétiques ont été développés pour faire face aux résistances acquises (oritavancine, télavancine) ou en raison de propriétés pharmacocinétiques permettant une administration hebdomadaire (dalbavancine). L’utilisation de ces molécules reste encore confidentielle en France. La résistance à la vancomycine est exceptionnelle chez S. aureus, même si des élévations de concentration minimale inhibitrice (CMI) jusqu’aux concentrations critiques rendent parfois possibles des échecs cliniques lors d’infections profondes. Des souches présentant une hétérorésistance (présence d’une sous-population de sensibilité diminuée au sein d’une population générale sensible à l’antibiotique) ont été décrites au début des années 2000 : elles ont été dénommées hétéro-GISA (glycopeptide-intermediate S. aureus). Par ailleurs une dizaine de souches de SARM résistant de haut niveau à la vancomycine et hébergeant le gène de la résistance aux glycopeptides des entérocoques ont été exceptionnellement rapportées. En effet chez ces derniers, la résistance à la vancomycine est portée par un gène mobile de la famille van ; les gènes vanA et vanB sont les plus fréquents. Les entérocoques résistants à la vancomycine (ERV) évoluent de façon épidémique en France contrairement aux États-Unis ou ils sont endémiques. La daptomycine (Figure 1F) est une molécule découverte dans les années 1980 à partir de cultures de Streptomyces roseosporus. Elle appartient à la famille des lipopeptides dont le mode d’action est lié à sa fixation à la membrane cellulaire de la bactérie qui entraîne sa dépolarisation rapide par action sur les canaux calciques puis l’inhibition de la synthèse de l’ADN, de l’ARN2 et des protéines. Sa toxicité neuromusculaire l’avait fait abandonner jusqu’au début des années 2000. L’émergence des souches hétéro-GISA et de souches présentant des CMI élevées aux glycopeptides a permis à la daptomycine de retrouver sa place au sein de l’arsenal des molécules ciblant les SARM. Son utilisation en une seule injection quotidienne a permis de limiter sa toxicité musculaire ; sa neutralisation par le surfactant pulmonaire l’empêche d’être utilisée lors d’infections pulmonaires. Sa toxicité rénale bien moins importante que celle des glycopeptides en fait une alternative très prisée. Son activité in vitro nécessite du calcium ionisé en quantité précise : l’antibiogramme pour tester cette molécule nécessite des conditions particulières de réalisation au laboratoire. Les souches de SARM ne sont qu’exceptionnellement résistantes à la daptomycine. Néanmoins, une co-résistance avec certains antiseptiques cationiques tels que les biguanides a été décrite dans la littérature et mérite d’être surveillée [8].
Les oxazolidinones dont le linézolide
Les oxazolidinones constituent la dernière nouvelle classe d’antibiotique découverte dans les années 2000 dont les représentants sur le marché sont le linézolide (Figure 1G) et le tédizolide. Ces molécules de synthèse inhibent le fonctionnement des ribosomes bactériens en se fixant sur la sous-unité 50S, bloquant la synthèse protéique ; ils sont efficaces sur les cocci à coloration de Gram positive comme les staphylocoques et les entérocoques, même lorsqu’ils sont résistants aux autres classes d’antibiotique. Ce sont donc des molécules de choix pour traiter les infections à SARM ou à ERV, d’autant qu’elles sont administrables par voie orale et par voie intraveineuse. Leur toxicité hématologique et neurologique (neuropathies) limite leur utilisation durant de longues périodes. Des résistances, chromosomiques ou dues à la présence d’éléments génétiques mobiles, ont été décrites ; leur diffusion est favorisée par une utilisation large de ces molécules et des dérives dans la prévention de la transmission croisée. Ces résistances restent épidémiques en France à ce jour. Un autre intérêt de ces molécules est leur capacité à inhiber la synthèse de toxines, ce qui peut être intéressant lors de certaines infections avec des souches toxinogènes de S. aureus ou de Streptococcus pyogenes (streptocoque du groupe A). Le linézolide est également utilisé pour le traitement de tuberculoses multirésistantes.
La colistine et les polymyxines
La colistine (Figure 1H) a été découverte en 1950. Elle est produite par certaines souches de Bacillus polymyxa var. colistinus. Elle appartient à la famille des polymyxines qui sont des antibiotiques polypeptidiques cycliques. La colistine, ou polymyxine E, n’agit que sur les bacilles à coloration de Gram négative dont elle solubilise la membrane cellulaire bactérienne, ce qui entraîne un effet bactéricide. Son activité est liée à un effet polycationique désorganisant les groupes phosphates des lipopolysaccharides de la membrane des bacilles à Gram négatif. La colistine n’est pas absorbable par voie orale : elle peut encore être utilisée sous la forme de comprimés ou de solutions buvables dans de rares protocoles de décontamination digestive, notamment en hématologie clinique. Par voie intraveineuse, la colistine présente une toxicité rénale très importante qui a conduit à son abandon au profit de bêtalactamines dont l’activité anti-Gram négative a été progressivement élargie. L’émergence de souches d’entérobactéries multirésistantes aux antibiotiques et productrices de carbapénémase, d’Acinetobacter baumannii résistantes à l’imipénème ou de Pseudomonas aeruginosa multirésistantes a permis à la colistine de retrouver une place en thérapeutique humaine, le plus souvent en association avec d’autres antibiotiques de recours. Il existe des utilisations hors autorisation de mise sur le marché sous la forme d’aérosol chez les patients atteints d’infection respiratoire. Il existe des espèces d’entérobactéries naturellement résistantes aux polymyxines : Serratia sp., Proteus sp., Providencia sp., Morganella morganii. Au sein des bacilles à Gram négatif non fermentant, les espèces appartenant au genre Burkholderia sont également naturellement résistantes. Les polymyxines ont été pendant longtemps très utilisées en médecine vétérinaire sous différentes formes d’administration. Le développement de l’approche « Une seule santé » a incité les vétérinaires à restreindre drastiquement l’utilisation chez l’animal de ces antibiotiques de derniers recours dans l’espèce humaine. La résistance acquise à la colistine était initialement d’origine chromosomique, avec des mutations identifiées au sein de certains gènes impliqués dans la structure du lipopolysaccharide avec pour conséquence une abolition de la fixation de la colistine sur la paroi bactérienne. Au début des années 2010, la résistance plasmidique à la colistine via les gènes mobiles mcr3 a été rapportée chez des souches bactériennes isolées chez l’animal, probablement favorisée par l’utilisation historiquement très large de cette classe d’antibiotique. Initialement identifiés chez E. coli, ces gènes et ces plasmides ont ensuite été retrouvés chez de très nombreuses espèces de bacille à Gram négatif. Il est à noter que la détection au laboratoire d’une résistance acquise aux polymyxines n’est pas simple et nécessite la détermination des CMI par dilution en milieu liquide, méthode coûteuse et consommatrice de temps qui n’est pas réalisée en routine. Quelques publications ont mis en évidence une relation directe entre la résistance à la colistine et la diminution d’activité de la chlorhexidine [9]. Ce phénomène inquiétant semble ponctuel au regard notamment de la très large utilisation de cet antiseptique. Une vigilance reste néanmoins nécessaire vis-à-vis de ce phénomène redouté de co-résistance entre antibiotique et antiseptique.
Les macrolides et les molécules apparentées
Comme leur nom l’indique, les macrolides sont des grosses molécules (ou macrocycles) qui comportent de 14 à 16 atomes de carbone. Les premiers datent des années 1950 avec l’érythromycine (Figure 1I) qui a été isolée d’une bactérie du sol (Streptomyces erythraeus). Ces molécules inhibent la synthèse protéique par fixation sur la sous-unité 50S du ribosome bactérien. Elles ont plusieurs propriétés très intéressantes qui ont conduit à une utilisation importante et donc à l’émergence de nombreuses résistances. Les macrolides ont une activité contre les bactéries à coloration de Gram positive comme les staphylocoques et les streptocoques dont le pneumocoque : ils sont donc très prescrits lors d’infections oto-rhino-laryngologiques (ORL), cutanées ou respiratoires communautaires. Pour cette dernière indication, les macrolides possèdent également une activité sur les bactéries intracellulaires comme les Chlamydia, les mycoplasmes et les légionelles, ce qui constitue un intérêt majeur dans les pathologies associées qui ne répondent pas aux bêtalactamines. De plus les développements industriels ont permis la mise au point de molécules peu toxiques comme la spiramycine ou présentant une longue demi-vie permettant des durées de traitement courtes voire en une seule dose (azithromycine). Elles présentent néanmoins des interactions médicamenteuses par inhibition des cytochromes P450 qu’il faut surveiller en cas de co-administration avec d’autres médicaments à marge thérapeutique étroite métabolisés par cette voie d’élimination. En plus d’être indiquée pour les infections respiratoires, ORL et cutanées, l’azithromycine est une molécule centrale dans le traitement de certaines infections sexuellement transmissibles (urétrite à Chlamydia trachomatis et à gonocoque) ; la clarithromycine est également une molécule importante dans le traitement des infections à mycobactérie atypique (Mycobacterium avium complex). Hormis un manque d’efficacité naturel sur Haemophilus influenzae, bactérie fréquemment retrouvée lors d’infections ORL ou respiratoires, l’activité des macrolides est surtout restreinte du fait de résistances acquises dont le support est chromosomique (mutations ribosomales) ou présent sur des éléments génétiques mobiles (gènes erm4 ou mef5) retrouvés chez les bactéries à Gram positif. Ainsi la résistance aux macrolides est retrouvée chez les pneumocoques et chez S. aureus : 25% des souches invasives isolées en 2022 étaient résistantes à l’érythromycine [2,10]. Chez les bactéries intracellulaires, ce sont des mutations qui sont responsables de la résistance acquise chez un nombre croissant de souches de Mycoplasma pneumoniae ; leur détection nécessite des méthodes moléculaires car les antibiogrammes standard ne peuvent pas être utilisés pour ces espèces. Il est à noter que l’érythromycine possède un pouvoir d’accélération de la vidange gastrique qui est encore utilisé en réanimation dans cette indication chez certains patients.
Des molécules apparentées aux macrolides ont été développées avec des propriétés intéressantes. La clindamycine est le dernier représentant des lincosamides. C’est une molécule qui se fixe également sur la sous-unité ribosomale 50S. Elle possède une activité contre de nombreuses bactéries anaérobies. Elle est donc utilisée lors d’infections mixtes aéro-anaérobies ou dans des schémas d’antibioprophylaxie impliquant la sphère ORL, génitale ou digestive, souvent pour remplacer les bêtalactamines lorsque le patient est allergique. L’utilisation large de la clindamycine aux États-Unis aurait favorisé les infections à Clostridioides difficile. Comme pour les macrolides, les résistances acquises sont fréquentes à la fois chez les staphylocoques, les streptocoques et les bactéries anaérobies. La clindamycine inhibe la sécrétion de toxine : elle a été utilisée seule ou en association lors d’infections à S. aureus ou S. pyogenes avant la découverte du linézolide avec lequel elle partage cette propriété (voir supra). La pristinamycine est le seul représentant encore commercialisé de la famille des synergistines ou streptogramines. C’est une association de deux composés A et B, qui reste stable malgré la présence de gènes de résistance aux macrolides. C’est une molécule disponible quasi exclusivement en France qui garde son intérêt pour traiter certaines infections ORL, respiratoires ou cutanées. Les souches de S. aureus, de pneumocoque et de S. pyogenes résistantes à la pristinamycine restent exceptionnelles.
Les cyclines et les antibiotiques apparentés
Les cyclines (Figure 1J) sont des molécules très anciennes qui partagent de nombreuses propriétés avec les macrolides : une découverte dans les années 1950, une origine naturelle (les bactéries du genre Streptomyces), une action via l’inhibition du ribosome bactérien (en se fixant sur la sous-unité 30S), une administration par voie orale et par voie injectable, une activité à la fois sur les staphylocoques, les streptocoques et autres pyogènes et sur les bactéries intracellulaires, et des taux de résistances acquises élevés. La tétracycline est le chef de file de cette famille. Plusieurs molécules ont été commercialisées ; aujourd’hui la doxycycline et la minocycline sont les principaux composés utilisés contre les infections à bactérie intracellulaire, les infections cutanées (les cyclines sont actives sur Cutibacterium acnes, bactérie de la peau associée à l’acné) mais aussi contre certaines infections à Stenotrophomonas maltophilia. Récemment, les cyclines ont été recommandées en prophylaxie dans la prévention des infections sexuellement transmissibles à Chlamydia trachomatis. Les cyclines ont également une activité antiparasitaire et sont utilisées par exemple en prophylaxie antipaludéenne. Leur toxicité chez l’enfant limite leur utilisation dans cette classe d’âge. Plus récemment la tigécycline, qui appartient à la famille des glycylcyclines, a été développée et commercialisée pour le traitement de dernière ligne de certaines infections à entérobactérie multirésistante, staphylocoque, ou bactérie anaérobie (infection compliquée de la peau et des tissus mous hors infection du pied chez le patient diabétique, infection intra-abdominale compliquée). C’est une molécule injectable dont l’efficacité est imparfaite mais qui peut être intéressante lors de certaines impasses thérapeutiques. D’autres dérivés comme l’omadacycline et l’éravacycline ont été récemment commercialisés.
Autres molécules antibiotiques
Les nitrofuranes
La nitrofurantoïne est le seul représentant commercialisé de la famille des nitrofuranes. Elle agit par inhibition de plusieurs systèmes enzymatiques bactériens sur des espèces à Gram négatif et à Gram positif et se concentre dans les urines. Son spectre bactérien inclut Escherichia coli, Staphylococcus saprophyticus et Enterococcus faecalis, les principales bactéries responsables d’infection urinaire basse : elle est donc indiquée pour le traitement probabiliste des cystites aiguës à risque de complication [5]. Les résistances acquises sont identifiées chez moins de 1% des souches d’E. coli mais chez 40,4% des souches de K. pneumoniae et 24,7% des souches d’E. cloacae complex [2].
L’acide fusidique
L’acide fusidique est une molécule de structure stéroïdienne issue d’un champignon : Fusidium coccineum. Il bloque la synthèse protéique de la bactérie en se fixant sur le ribosome via le facteur G impliqué dans la translocation de la chaîne des peptides durant la synthèse des protéines. Son spectre est limité aux staphylocoques et aux streptocoques. L’acide fusidique a été très utilisé sous forme locale (pommade, crème) pour traiter des infections dermatologiques bénignes comme l’impétigo. Cette utilisation a eu pour conséquence l’émergence de la résistance acquise, ce qui est regrettable puisque cette molécule peut être utilisée par voie orale ou injectable en association avec d’autres antibiotiques pour traiter des infections ostéo-articulaires à SARM. Néanmoins, en 2022, 14,3% des souches de SARM étaient résistantes à l’acide fusidique [2].
La mupirocine
La mupirocine est produite par Pseudomonas fluorescens : c’est le seul représentant de cette classe d’antibiotique qui possède une structure et un mécanisme d’action uniques. La mupirocine inhibe la synthèse protéique bactérienne par liaison à l’isoleucyl-ARNt6 synthétase. Elle n’est disponible qu’en pommade, indiquée dans le traitement des infections cutanées non sévères comme les impétigos et pour la décolonisation nasale à S. aureus. Les résistances acquises existent par mutation chromosomique ou par acquisition de gènes additionnels mobiles ; elles sont de bas ou de haut niveau, seul ce dernier étant à l’origine d’échecs cliniques. Néanmoins, une combinaison de résistances acquises associant une résistance de bas niveau à la mupirocine et la présence de gènes associés à une diminution de sensibilité à la chlorhexidine a été rapportée comme associée à l’échec de la décontamination cutanéomuqueuse de S. aureus [11].
La rifampicine et ses dérivés
La rifampicine est un antibiotique de la famille des rifamycines. Elle a été développée en 1968 à partir de la rifamycine, antibiotique produit par Streptomyces mediterranei. Son spectre antibactérien inclut les staphylocoques, les streptocoques et les bactéries à croissance intracellulaire telles que les mycobactéries, les brucelles, les Coxiella… La rifampicine agit en inhibant l’ARN polymérase des bactéries, empêchant ainsi la transcription de leur ADN et donc leur réplication. La rifampicine est un des seuls antibiotiques qui agissent sur les bactéries quiescentes, c’est-à-dire qui ne sont pas en phase de multiplication. Cela en fait une molécule particulièrement intéressante lors d’infections incluant des bactéries ou des souches se multipliant très lentement, au sein de biofilms par exemple. La résistance acquise à la rifampicine survient très rapidement : il ne faut donc pas utiliser cette molécule par voie générale sans l’associer à un autre antibiotique. La rifamycine peut être utilisée sous une forme locale (collyre). La rifampicine est utilisée en association à d’autres antibiotiques lors d’infections sévères à staphylocoque ou streptocoque (infections ostéo-articulaires, endocardites…). Avec l’isoniazide, la rifampicine constitue le pilier du traitement de la tuberculose. La prévalence de la résistance primaire (c’est-à-dire avant tout traitement antibiotique) des souches de Mycobacterium tuberculosis varie selon les pays : en France elle est inférieure à 2% [12]. D’autres dérivés de la rifampicine sont utilisés en pathologie humaine dans des situations cliniques différentes : la rifabutine est prescrite en association pour traiter les infections à mycobactérie atypique alors que la rifaximine, dérivé peu absorbable, est utilisée chez les patients cirrhotiques pour modifier leur microbiote intestinal et diminuer la fabrication d’ammoniaque à partir d’urée, dans le but de prévenir l’encéphalopathie hépatique.
Les nitro-5-imidazolés
Les nitro-5-imidazolés (métronidazole, secnidazole, tinidazole…) sont les antibiotiques de référence pour le traitement des infections à bactérie anaérobie, à quelques exceptions près comme Cutibacterium acnes. Ils n’ont pas d’activité sur les autres bactéries. Ce sont des molécules bien absorbées par voie orale, mais aussi administrables par voie intraveineuse. Elles sont indiquées contre les infections dues à des bactéries anaérobies (infections digestives, ORL, génitales…) en association avec d’autres molécules agissant sur les entérobactéries et les entérocoques. Les résistances acquises se sont développées, notamment au sein du genre Bacteroides. Les nitro-5-imidazolés sont également utilisés en prophylaxie lors d’interventions chirurgicales, associés à un aminoside par voie orale par exemple lors d’une chirurgie colorectale [4]. Les nitro-5-imidazolés possèdent d’autres activités dont une action antiparasitaire sur les protozoaires intestinaux ou génitaux (Giardia intestinalis, Entamoeba histolytica, Trichomonas vaginalis…) et une action anti-inflammatoire locale utilisée notamment dans le traitement de la rosacée.
Conclusion
Ce second article clôture cette vision globale de notre arsenal thérapeutique antibiotique. Il est remarquable de noter que cet arsenal est très majoritairement basé sur des molécules qui ont été découvertes il y a quarante, cinquante voire plus de quatre-vingts ans. La mise en évidence de nouvelles classes thérapeutiques aboutissant à des médicaments commercialisés reste exceptionnelle, la dernière datant d’il y a plus de vingt ans. En parallèle de cet épuisement du « pipeline » des antibiotiques, les résistances acquises se sont systématiquement développées, précédant parfois l’utilisation à l’échelle humaine de la molécule en raison de l’origine très majoritairement naturelle de ces composés. L’industrie pharmaceutique s’est attachée à améliorer chimiquement les molécules princeps sur le plan de la tolérance, de la biodisponibilité et du spectre antibactérien. Étonnamment, même lorsque des antibiotiques de synthèse ont été produits, des résistances acquises se sont développées sur un mode mutationnel attendu, mais également par l’intermédiaire de l’acquisition de gènes additionnels retrouvés sur des éléments génétiques mobiles passant d’une souche à l’autre, voire d’une espèce bactérienne à une autre. D’autres éléments sont inquiétants, comme l’élargissement du spectre d’hydrolyse d’une enzyme entraînant l’élargissement de la résistance à une autre famille de molécules, la coexistence sur un même élément génétique mobile de gènes de résistance à de nombreuses familles d’antibiotique et l’association de la résistance aux antibiotiques à la diminution de la sensibilité aux antiseptiques. La présence de ces phénomènes de résistance acquise chez les champignons (souches de Candida auris multirésistantes, ou d’Aspergillus résistantes aux azolés) et parasites (Plasmodium résistant aux antipaludéens) souligne leur caractère inexorable. Le développement des techniques de séquençage à haut débit des génomes microbiens et de criblage de composés chimiques, associé aux outils d’intelligence artificielle, laisse espérer la découverte de nouvelles molécules. Néanmoins, une expérience de bientôt cent ans nous assure que rien ne remplacera la prévention, qu’elle soit vaccinale ou surtout non médicamenteuse comme nous la pratiquons au quotidien au sein des équipes de prévention du risque infectieux.
Notes :
1- Acide désoxyribonucléique.
2- Acide ribonucléique.
3- Mobilized colistin resistance.
4- Erythromycin resistant methylase.
5- Macrolide efflux.
6- ARNt : ARN de transfert.