Principe du typage microbien
Le typage microbien est la caractérisation de sous-populations au sein d’une espèce. L’espèce est un groupe de micro-organismes qui présentent une homogénéité génétique stable dans le temps. Au sein de cette espèce, chaque individu peut évoluer génétiquement au cours du temps. Il acquiert alors des mutations et autres changements de son matériel génétique (délétion, insertion de gènes…). Cette évolution aboutit à une diversité génétique au sein d’une même espèce. L’étude de cette diversité génétique et l’articulation des différences sous-populations entre elles s’appelle la phylogénie. C’est grâce à cette évolution génétique au sein d’une espèce qu’il est possible de typer et de comparer des souches microbiennes au cours d’une épidémie, qui correspond à l’identification regroupée dans le temps et dans l’espace d’au moins deux cas de colonisation ou d’infection par le même agent infectieux [2].
Le principe de la comparaison des souches est le suivant : si les souches impliquées dans le phénomène épidémique ont un lien de parenté élevé, elles diffèrent génétiquement entre elles significativement moins qu’avec d’autres souches avec lesquelles elles n’ont que peu ou pas de lien de parenté. La durée de l’épidémie peut être à l’origine de modifications au sein des souches impliquées, même si elles ont une origine génétique commune ; ces modifications sont néanmoins bien moins nombreuses que celles observées avec des souches non reliées génétiquement qui ont évolué séparément depuis des années. À titre d’exemple, pour une bactérie, la fréquence spontanée de mutation (c’est-à-dire la substitution d’un nucléotide par un autre) sur chaque position de l’ADN2 varie entre 10-5 et 10-8 par an. Cette fréquence des substitutions nucléotidiques varie sensiblement en fonction de l’espèce : elle est ainsi plus élevée chez Acinetobacter baumannii ou Enterococcus faecium que chez Klebsiella pneumoniae ou Mycobacterium tuberculosis. Pour un génome de 5 millions de paires de bases, cela correspond à une échelle de 0,5 à 25 substitutions par an, soit entre 0 et 2,1 substitutions donc mutations par mois [3]. Pour le SARS-CoV-2, cette fréquence de mutations spontanées a été évaluée entre 0 et 2 par mois [4]. C’est le « bruit de fond » de l’évolution génétique dont il faut tenir compte lorsque l’on compare deux souches suspectées d’avoir un lien de parenté fort. D’autres paramètres peuvent avoir un impact sur ce bruit de fond : la pression de sélection par des anti-infectieux (antibiotiques, antiseptiques, désinfectants…), par l’immunité acquise (réponse immunitaire incluant l’utilisation des vaccins) ou par le fond génétique de certaines souches dites « hyper-mutatrices », c’est-à-dire qui génèrent spontanément un grand nombre de mutations, par exemple par défaut de correction des erreurs de transcriptions.
Place de la comparaison de souches microbiennes : déchiffrer les chaînes de transmission
La transmission des micro-organismes est basée sur le même principe immuable : l’exposition d’un hôte réceptif à un micro-organisme présent dans un réservoir, puis son acquisition par une voie ou plusieurs voies de transmission (contact, gouttelettes, aérosol, aliment, environnement, vecteur vivant…). L’hôte devient colonisé et, parfois, infecté. Sous certaines conditions, il peut lui-même devenir un réservoir (Figure 1). En milieu hospitalier, une épidémie telle que définie précédemment peut être due à des phénomènes de transmissions croisées entre patients ou à l’exposition de plusieurs patients au même réservoir (matériel contaminé, réservoir environnemental, aliment…). Le rôle de l’hygiéniste est d’enquêter et d’émettre des hypothèses sur ces réservoirs et voies de transmissions. Il va réaliser des prélèvements pour explorer certains maillons de cette chaîne de transmission : dépistage de patients « contact » ou « source », prélèvement de réservoirs primaires, sources des micro-organismes, ou secondaires. Il pourra ainsi mettre en place des mesures correctives : élimination des réservoirs (matériel ou circuits d’eau contaminés) ou à défaut leur maîtrise (isolement des patients porteurs), cassure des voies de transmission par des mesures adaptées (renforcement de l’hygiène des mains, port des masques et autres matériels de protection adaptés…). Pour déchiffrer ces chaînes de transmission, l’hygiéniste doit séparer les souches impliquées dans l’épidémie de celles présentes de façon aléatoire. Cela est surtout important lorsque le micro-organisme circule de façon naturelle indépendamment de l’épidémie investiguée : circulation communautaire de virus grippaux ou de la Covid-193, bruit de fond de portage d’une bactérie multirésistante… Pour cela, il doit établir les caractéristiques des différentes souches identifiées et les comparer.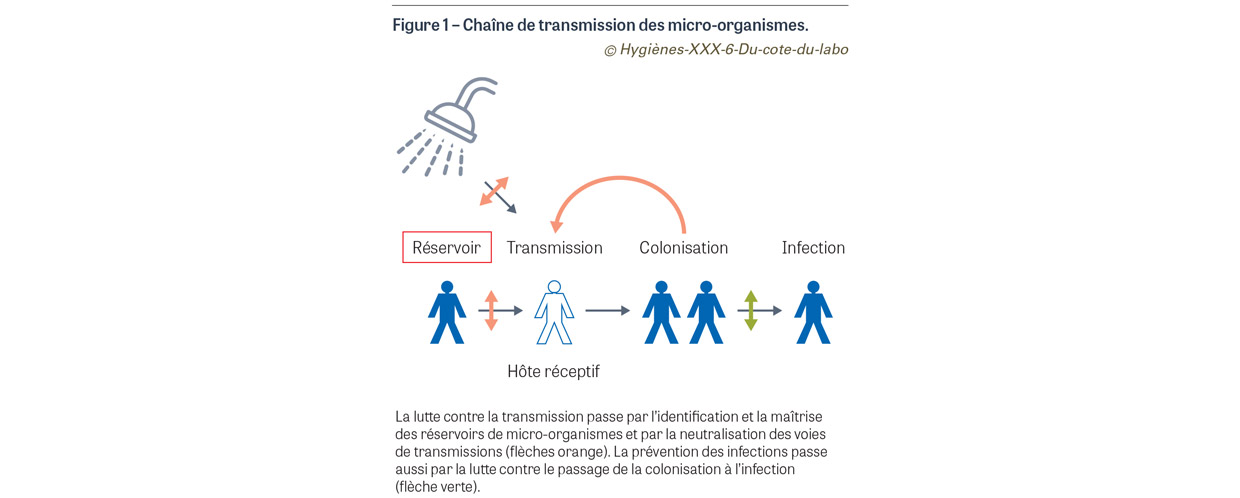
La comparaison hier
L’objectif est donc de caractériser des souches pour voir si elles présentent des différences entre elles, et d’établir si ces différences sont mineures et correspondent à leur évolution spontanée au cours de l’épidémie, ou si elles sont tellement nombreuses qu’elles sont dues à un lien de parenté trop éloigné pour qu’elles soient considérées comme liées dans le cadre du phénomène épidémique étudié. L’approche la plus simple est de comparer les caractéristiques phénotypiques, qui ne sont que l’expression apparente des caractéristiques génétiques. Pour les bactéries et les champignons, l’aspect des colonies en culture, le profil de sensibilité aux anti-infectieux (appelé « antibiotype ») ou certains caractères biochimiques sont des méthodes anciennes, dont certaines sont toujours utiles au moins pour donner l’alerte : un profil de sensibilité aux antibiotiques atypique ou particulièrement résistant partagé par plusieurs souches alors qu’il est rarement rencontré est un signe d’alerte d’un phénomène épidémique. La sensibilité aux phages (lysotypie) et les spécificités enzymatiques et métaboliques ne sont désormais plus utilisées. La sérotypie qui utilisait des antisérums permettant de mettre en évidence des profils antigéniques particuliers, notamment polysaccharidiques, a aussi été progressivement abandonnée : elle a connu son heure de gloire notamment pour typer les souches de Pseudomonas aeruginosa. Le coût des antisérums était inadapté aux performances somme toute relatives de ce typage. Depuis une trentaine d’années, des approches génotypiques ont été développées, notamment pour mettre en évidence des différences de composition génétique [5,6]. La restriction enzymatique permettait de rechercher des sites de restrictions particuliers à chaque enzyme et caractéristiques de chaque souche. La méthode qui a été longtemps considérée comme la référence était ainsi fondée sur la restriction enzymatique puis la migration des fragments obtenus par une électrophorèse adaptée aux gros fragments d’ADN : la comparaison des profils de macro-restriction par électrophorèse en champ pulsé (Pulsed-field gel electrophoresis [PFGE]). Cette méthode était longue (5 jours), très consommatrice de temps technique, et coûteuse en raison du prix élevé des enzymes de restriction et de la nécessité de disposer d’un matériel d’analyse dédié. Les critères d’interprétation basés sur le nombre de bandes différentes après électrophorèse étaient sommaires mais reconnus internationalement (Figure 2A). Des trousses prêtes à l’emploi ont été commercialisées et des réseaux de surveillance nationaux ont été basés sur cette approche après la mise en place d’une uniformisation des protocoles. La seconde grande approche a été l’utilisation des techniques d’amplification génétique par réaction de polymérisation en chaîne (Polymerase chain reaction [PCR]) qui mettaient en évidence des différences génétiques en utilisant des amorces ciblant des régions intergéniques de taille variable ou des amorces peu spécifiques s’hybridant et amplifiant des séquences de façon aléatoire et peu spécifique. Ces techniques avaient l’avantage d’être peu coûteuses et réalisables dans de nombreux laboratoires sans matériel dédié. Leur interprétation était néanmoins aléatoire, et les résultats étaient peu reproductibles et impossibles à échanger entre laboratoires. Une trousse prête à l’emploi avec du matériel dédié basé sur la technique de la rep-PCR4 a longtemps été commercialisée ; le laboratoire a arrêté sa production il y a quelques années au profit de solutions de séquençage de génomes complets. La dernière approche développée et qui est toujours d’actualité est basée sur le séquençage d’un ou plusieurs gènes. Les avantages sont nombreux : des séquenceurs nombreux donc disponibles qui peuvent être partagés entre différents secteurs (microbiologie, génétique humaine…), des données de séquence pouvant être facilement échangées entre laboratoires et partagées sur des plateformes, et une nomenclature internationale facilement accessible en ligne. Initialement, le séquençage a concerné un gène (par exemple : le gène spa pour Staphylococcus aureus) : à chaque séquence est attribué un chiffre correspondant à un type particulier. Cette technique a également été et est toujours utilisée pour typer les virus. Pour augmenter la puissance de discrimination de la technique, le séquençage a été étendu à 5 voire 7 gènes dits « de ménage » (« housekeeping genes ») toujours présents dans les espèces bactériennes concernées. À chaque séquence correspondant à un allèle particulier est attribué un numéro ; le code correspondant à l’ensemble des différents gènes, donc des allèles, permet d’attribuer un numéro de type. Cette technique, dite Multi-locus sequence typing5 (MLST), est encore très utilisée pour étudier la distribution et la diffusion de certaines sous-populations au niveau national ou mondial6. Son intérêt dans l’investigation relative à un phénomène épidémique peut être limité si un type de séquence est majoritairement présent dans un pays car la technique n’est pas assez discriminante.
La comparaison aujourd’hui
Comparaison de bactéries
Les techniques phénotypiques se limitent désormais à l’antibiotype, caractéristique importante pour la détection des épidémies de bactéries multirésistantes. La démocratisation et la mise en place dans la quasi-totalité des laboratoires de la technique d’identification des bactéries et des levures par spectrométrie de masse de type MALDI-TOF (Matrix assisted laser desorption ionization - Time of flight7) laissait espérer une utilisation des profils protéiques obtenus pour comparer les souches ; à ce jour, pourtant, aucun protocole standardisé n’existe. Depuis quelques années, une solution commercialisée avec matériel et réactifs dédiés permet une comparaison rapide en quelques minutes des profils d’absorption des sucres des colonies bactériennes et des levures dans le spectre infra-rouge. La théorie qui sous-tend cette technique basée sur la spectrométrie infrarouge à transformée de Fourier (IRTF) a été mise au point il y a plusieurs années [7,8,9]. La mise à disposition de logiciels permettant d’interpréter les spectres d’absorption a donné une nouvelle jeunesse à cette approche. L’appareillage est dédié et coûteux (environ 70 000 euros) mais le coût des réactifs rapporté à la souche est de quelques euros. Les conditions de culture des différentes souches avant comparaison doivent être parfaitement maîtrisées : type de milieu, durée de la culture, hygrométrie de l’environnement… Cela oblige à un repiquage
systématique des différentes souches à tester, donc à un délai supplémentaire de 18 heures avant la comparaison, qui ne dure que quelques minutes. Les critères d’interprétation varient pour chaque acquisition de spectre, ce qui rend l’interprétation parfois difficile. Les représentations des résultats dans l’espace par les différentes fonctions du logiciel permettent néanmoins une visualisation assez simple des liens de parenté entre les différentes souches, permettant d’exclure, à défaut de confirmer, ces liens de parenté (Figure 2B). Cela pourrait être une technique de première ligne, rapide, permettant d’avoir des résultats préliminaires avant le déclenchement d’une technique de référence, plus longue et plus coûteuse. Les techniques basées sur la séquence d’un ou plusieurs gènes sont toujours d’actualité, mais leurs performances sont aujourd’hui dépassées par le séquençage des génomes complets [1,5,6]. Utilisant les séquenceurs à haut débit qui équipent désormais toutes les plateformes de génomique, le séquençage du génome complet d’une bactérie ne coûte aujourd’hui que quelques dizaines d’euros. L’accès à l’ensemble de l’information génétique de chaque souche bactérienne permet de les comparer de manière performante : les mutations peuvent être recherchées sur tout ou partie du génome (analyse des single nucleotide polymorphisms8) ou une approche par typage MLST peut être développée en ne ciblant plus 5 à 7 gènes mais plusieurs centaines voire milliers de gènes en commun (core-genome MLST) [10]. Différents logiciels commerciaux sont disponibles, simplifiant l’analyse des données sans que l’utilisateur ait à disposer de compétences bio-informatiques particulières. Les différences et distances génétiques sont ensuite modélisées sur des arbres phylogéniques (Figure 2C).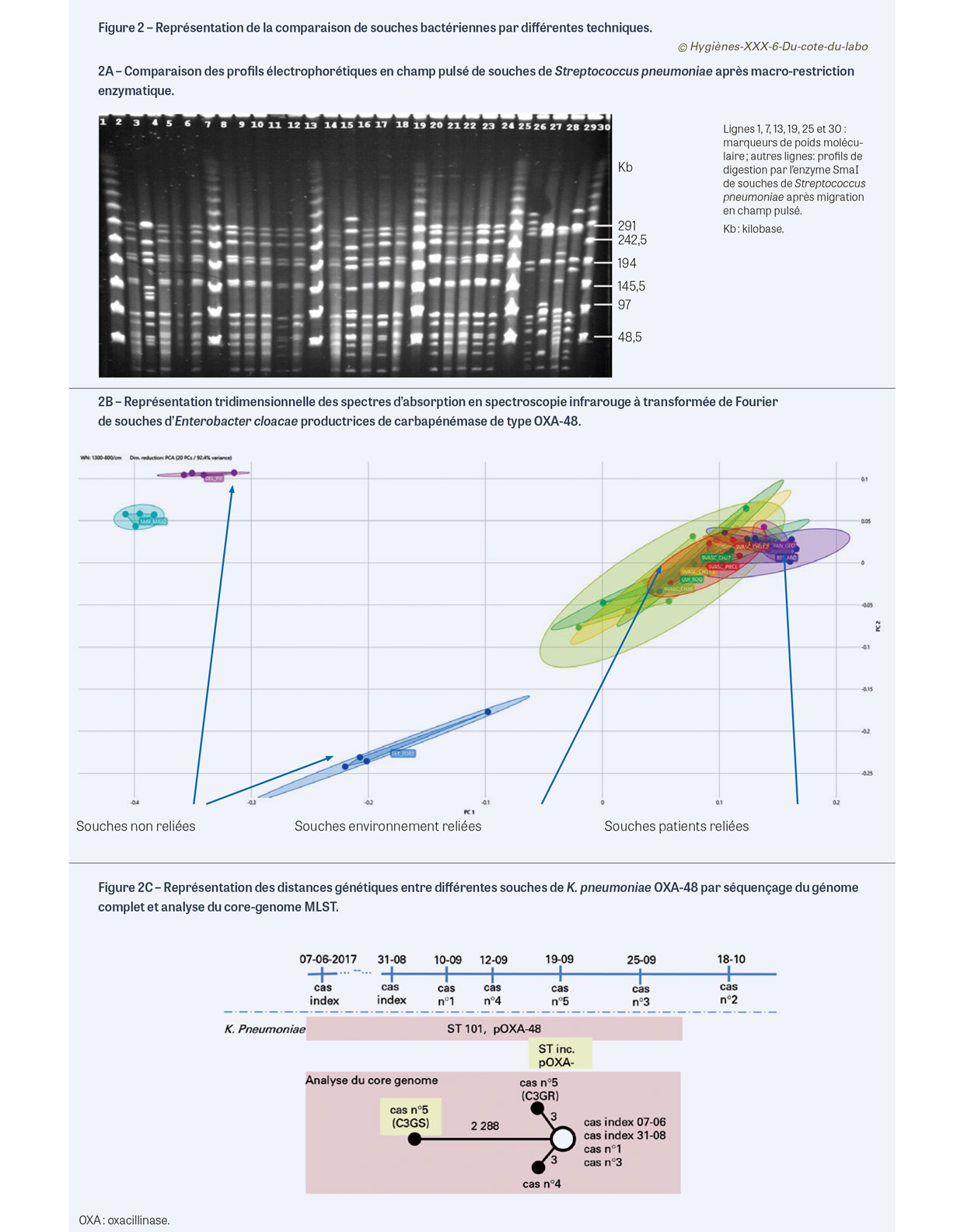
Comparaison de virus
La comparaison des virus reste basée sur le séquençage de tout ou partie du génome. Les logiciels commercialisés évoqués précédemment incluent des options pour pouvoir utiliser les données du séquençage complet. La taille des génomes et l’approche ciblant un gène précis rendent les techniques de comparaison plus simples, en utilisant les logiciels d’alignement des séquences habituellement disponibles en biologie moléculaire. À titre d’information, la taille du génome du SARS-CoV-2 et du virus de la grippe est respectivement de l’ordre de 30 000 et 15 000 nucléotides, alors que celle du génome d’une bactérie varie entre 2 et 6 millions de nucléotides. Le séquençage du génome complet des virus et l’utilisation de techniques bio-informatiques puissantes permettent la comparaison d’un grand nombre de souches en même temps et l’identification de voies de transmission au niveau d’un hôpital ou d’une ville [11,12].
Comparaison de champignons et de parasites
En termes de taille de génome, les champignons et les parasites appartiennent à un autre univers : l’information génétique est compactée sous la forme de chromosomes, qui sont parfois en double exemplaire (organismes eucaryotes). La taille du génome de Candida albicans est ainsi de 16 millions de nucléotides, celle du génome d’Aspergillus fumigatus, de 30 millions de nucléotides. La comparaison de différentes souches de champignons filamenteux utilise encore les techniques d’amplification des zones de polymorphisme (technique des microsatellites). Les techniques de séquençage restent ciblées sur des gènes précis car la taille des génomes demeure un écueil pour les approches de séquençage de génomes complets. La réalisation et l’interprétation des résultats relèvent encore à ce jour des centres de références.
Performances analytiques et limites de l’approche
Concernant les bactéries, la comparaison des souches par séquençage du génome complet constitue aujourd’hui la méthode de référence, facilement accessible sur les plateformes de séquençage ou par l’intermédiaire d’un prestataire externe. Bien plus performantes que les techniques historiques qui pouvaient être non pertinentes en cas de prédominance hégémonique d’une sous-population dans une région donnée, elles permettent dans la plupart des cas de répondre à la question du lien de parenté compatible avec une origine commune lors d’une épidémie. Il existe néanmoins plusieurs écueils :
- la qualité du séquençage doit être suffisante pour que les données de comparaison soient interprétables. L’hygiéniste doit être capable d’évaluer la qualité de l’information qui lui est donnée avant d’utiliser ces données pour prendre des décisions parfois lourdes de conséquences (arrêt des admissions et des transferts, fermeture de lits…) ;
- les délais de séquençage sont d’environ 7 jours et ceux de l’analyse bio-informatique de quelques jours supplémentaires, soit un délai total de 10 jours en moyenne ;
- dans les situations où l’interprétation est moins évidente (présence d’un nombre limité mais non nul de différences entre deux souches), les critères pour mesurer le degré de parenté ne font pas aujourd’hui l’objet de recommandations nationales ou internationales établies. Un travail collaboratif entre la Société française d’hygiène hospitalière et la Société française de microbiologie est en cours pour produire un guide technique à destination des hygiénistes et des microbiologistes ;
- l’un des principaux défis actuels que doivent relever les hygiénistes est la maîtrise de la diffusion des entérobactéries productrices de carbapénémases (EPC). Le support génétique de la résistance est le plus souvent plasmidique : il est donc possible d’avoir des épidémies de plasmides en plus des épidémies de souches. Cela est particulièrement vrai pour les EPC appartenant à la famille des OXA-489, famille des carbapénémases la plus fréquente en France. Les techniques les plus courantes de séquençage de génomes complets ne sont pas assez performantes pour reconstruire intégralement les plasmides des souches testées et les comparer. Il faut utiliser en complément des techniques plus puissantes, beaucoup plus chères et qui nécessitent des analyses bio-informatiques plus pointues ;
- concernant les épidémies de champignons filamenteux, il faut faire face à des difficultés de performance et d’interprétation liées à une très grande diversité de la contamination environnementale. Retrouver l’origine environnementale d’une contamination fongique est une requête particulièrement difficile et aléatoire.
Intérêt selon l’acteur
Intérêt pour le biologiste
La comparaison de souches est une demande qui est faite assez fréquemment au biologiste par les hygiénistes et les cliniciens. Le biologiste est par ailleurs lui-même à l’origine de la demande lorsqu’il détecte un phénotype de résistance particulier partagé par plusieurs isolats de patients appartenant à un même service. Disposer d’une technique rapide pour éliminer rapidement les fausses épidémies et d’une technique de référence pour au contraire confirmer un phénomène émergent fait partie des compétences que l’on demande à un laboratoire de qualité. La place du biologiste et ses interactions avec les plateformes de séquençage sont variables : il peut sous-traiter l’ensemble de l’activité en y incluant l’interprétation des données ou au contraire développer son expertise à partir des données brutes fournies. Le biologiste doit être impliqué dans la bonne gestion de l’épidémie : confirmée ou infirmée trop tardivement, elle peut avoir pour conséquence l’arrivée massive et parfois inutile de nombreux prélèvements supplémentaires de dépistage dans son laboratoire.
Intérêt pour le clinicien
La bonne gestion des épidémies est importante pour le clinicien : responsable de la qualité des soins qu’il prodigue à ses patients, il doit identifier et stopper rapidement les transmissions croisées de micro-organismes dans son service. De plus, le fonctionnement de ce service peut être perturbé par la prise en charge de l’épidémie, l’arrêt des admissions et des transferts paralysant son activité tant que le phénomène épidémique n’est pas compris et maîtrisé. La comparaison des souches par séquençage des génomes complets peut également lui apporter d’autres informations utiles : lors d’infections rapprochées chez un patient avec une même espèce, elle permet de faire la différence entre une réinfection avec une nouvelle souche ou une rechute avec la même souche initiale, deux phénomènes physiopathologiques qui n’ont pas les mêmes conséquences thérapeutiques. La disponibilité de l’ensemble du patrimoine génétique de la souche permet d’avoir accès à l’ensemble des gènes de résistance et de virulence. Ces informations peuvent être intéressantes pour compléter l’antibiogramme et aider à prendre certaines décisions thérapeutiques.
Intérêt pour le patient
Comme nous venons de le voir, le patient peut bénéficier individuellement des résultats de la comparaison de souches pour le choix de son traitement. Par ailleurs, l’identification rapide et sûre des différents paramètres d’une épidémie (réservoir, voie de transmission) permet de limiter rapidement son risque d’exposition. Enfin, dans le cadre d’un contexte médico-légal, cette comparaison constitue la preuve ultime d’une infection exogène dont l’évitabilité peut alors être discutée.
Impact pour l’hygiéniste
Aujourd’hui
Comme évoqué précédemment, la comparaison des souches microbiennes constitue un outil important pour l’hygiéniste. Elle lui permet de confirmer que des souches qui présentaient des caractéristiques similaires avaient bien une origine phylogénique commune. Ces caractéristiques peuvent être un phénotype particulier de résistance aux antibiotiques, une date ou un lieu d’isolement proches. Cette appartenance à un même cluster « temporo-spatial » sous-entend un mécanisme de transmission croisée ou d’exposition à un même réservoir. L’identification de cette voie de transmission est l’essence même du travail d’enquête de l’hygiéniste. Une fois que son existence est confirmée par des méthodes scientifiques dont les résultats sont indiscutables, l’hygiéniste peut poursuivre son travail d’investigation pour comprendre les mécanismes fins de transmission : voies de transmission (mains, gouttelettes, surfaces, matériel partagé…), existence d’un réservoir primaire ou secondaire, mesures de prévention qui ont fait défaut (désinfection, hygiène des mains…). Il peut ensuite mettre en place des actions correctives et en vérifier l’efficacité : l’absence de nouveau cas génétiquement lié atteste de la pertinence et de l’observance des mesures. Lorsque l’événement impliqué est rare, l’identification d’un nouveau cas est assez probablement rattachée à l’événement en cours (par exemple isolement d’une nouvelle souche d’une bactérie présentant un même phénotype de résistance rarement identifié chez un patient contact dépisté) : la contribution de la confirmation par les techniques de comparaison est relativement faible. À l’inverse, lorsque l’événement est fréquent en dehors de l’épisode épidémique (par exemple cas de Covid-19 au sein d’un cluster alors que l’incidence dans la communauté est forte), il est essentiel de confirmer l’appartenance de la souche isolée au cluster avant de remettre en cause les mesures mises en place pour contenir celui-ci. Lors de l’investigation relative à un cluster nosocomial de Covid-19 dans un service de long séjour gériatrique, nous avons pu exclure un cas identifié lors des dernières séries transversales de dépistage grâce à une approche par séquençage des souches de SARS-CoV-2, ce qui nous a permis de ne pas durcir plus que de raison les mesures coercitives mises en place dans le service concerné [4].
Une autre contribution des techniques de comparaison est de pouvoir convaincre les collègues cliniciens et paramédicaux de terrain de la véracité de l’épidémie identifiée par les hygiénistes ; souvent dubitatifs, rarement de mauvaise foi, nos collègues ont parfois besoin de la preuve « ultime » avant d’accepter l’existence d’une épidémie possiblement en lien avec un dysfonctionnement dans leur service. Au regard de cette place cruciale dans la prise de décision, l’hygiéniste gagnerait à pouvoir disposer d’une analyse critique des résultats de la comparaison de souches : qualité du séquençage, critères d’interprétation utilisés, pertinence des résultats… L’inconvénient majeur pour l’hygiéniste est que ces techniques ne sont utilisées que pour confirmer un phénomène épidémique préalablement suspecté. En l’absence de ces signes d’appel, les comparaisons de souches ne sont pas effectuées donc les liens phylogéniques ne sont pas identifiés. C’est la limite de l’approche épidémiologique historique, dite de John Snow, qui avait permis de découvrir le rôle de la contamination de certains puits lors de l’épidémie de choléra qui était survenue à Londres en 1854. John Snow avait en effet recoupé les lieux d’habitation des patients atteints avec la localisation des puits où la population venait se ravitailler [13]. Il n’avait pas à l’époque à disposition des méthodes de comparaisons de souches. C’est encore ainsi que nous travaillons aujourd’hui : lors d’une alerte émise par le laboratoire ou un service de soins, nous effectuons une enquête temporo-spatiale, émettons des hypothèses que nous confirmons en comparant les souches impliquées (Figure 3A). Pour pallier cette limite, certains hygiénistes et épidémiologistes ont développé une nouvelle approche dans laquelle la comparaison des souches n’est plus l’aboutissement d’une investigation, mais son début.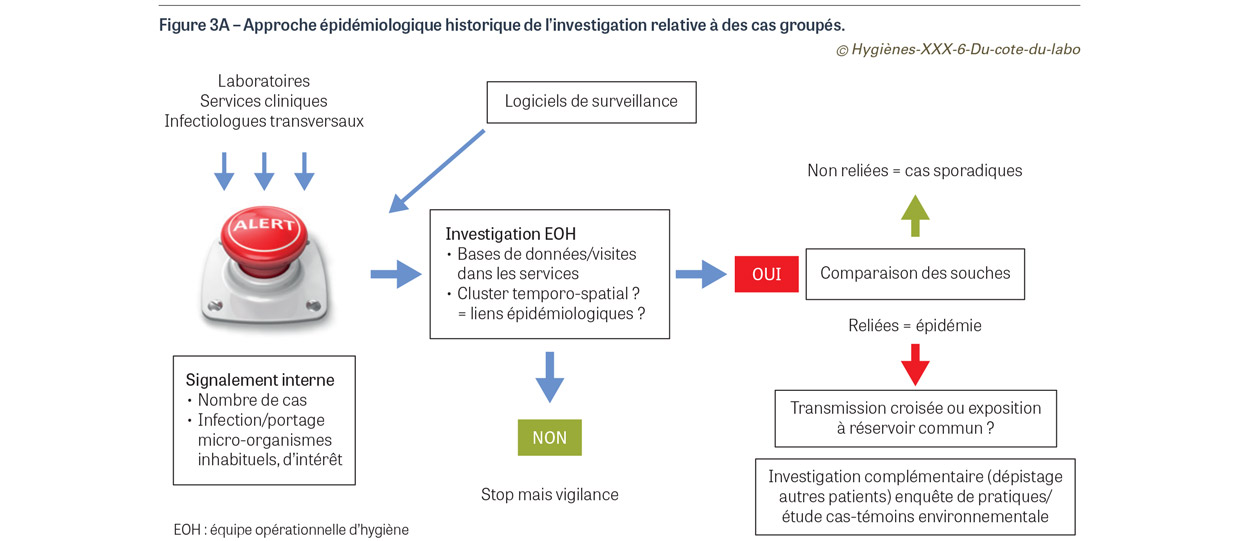
Demain : la comparaison de souches comme point de départ de l’investigation épidémique
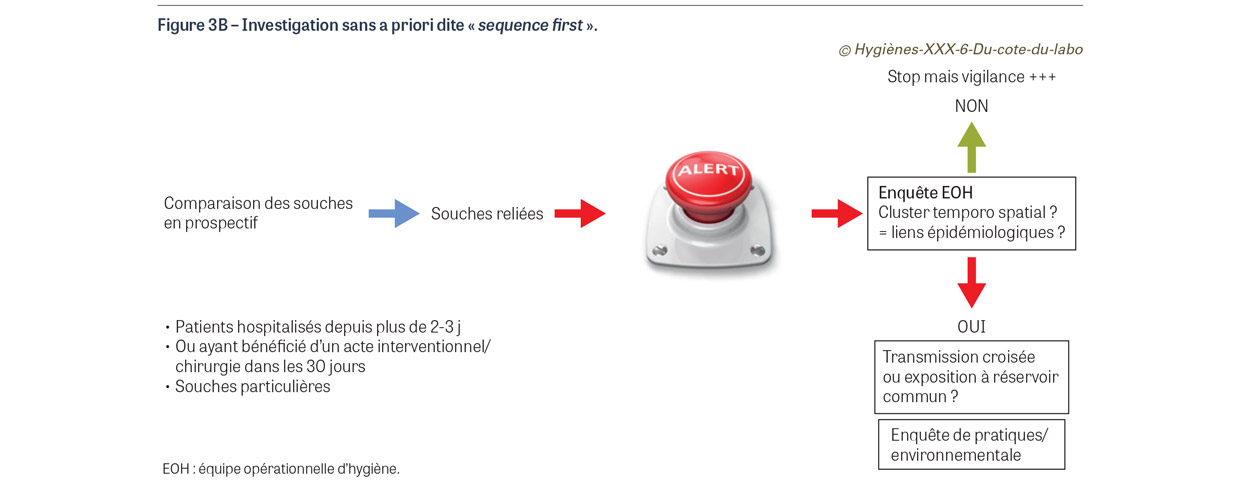
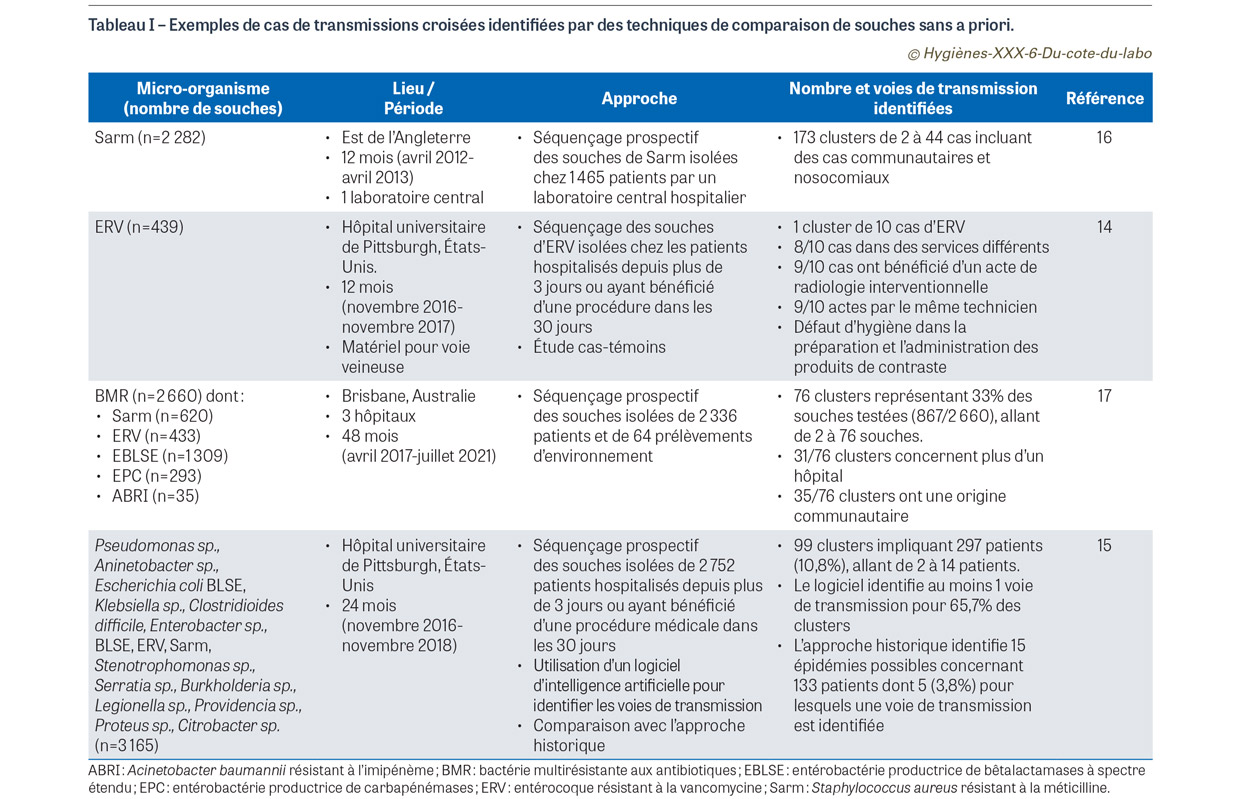
Cette approche s’est développée et fait connaître sous l’appellation « sequence first » avec l’avènement du séquençage de nouvelle génération qui a diminué de façon drastique les coûts humains et matériels de la comparaison de souches, en en raccourcissant le délai et en en facilitant l’interprétation [13]. Les souches microbiennes préalablement ciblées comme prioritaires sont séquencées de façon systématique sans présélection épidémiologique. C’est une approche « sans a priori ». Si une parenté génétique est identifiée, l’alerte est lancée et l’enquête épidémiologique déclenchée (Figure 3B). Cette approche avait déjà été développée avant l’avènement des techniques de séquençage à haut débit par certains centres de référence. Ainsi, le réseau Pulsenet des CDC10 aux États-Unis comparait, sans a priori, les profils de macro-restriction en champ pulsé des souches de salmonelles isolées, identifiant ainsi des parentés génétiques qui déclenchaient une enquête alimentaire11. Les difficultés de reproductibilité et d’échange des données liées à cette technique avaient tant bien que mal été lissées par la mise en place d’un protocole rigoureux. Ce réseau perdure mais la technique utilisée est maintenant le séquençage des génomes complets. Cette approche présente l’avantage de pouvoir identifier des événements dont les liens épidémiologiques ne sont pas évidents : dans l’étude de Sundermann et al. concernant une épidémie d’entérocoques résistants à la vancomycine, la quasi-totalité des patients infectés avaient été hospitalisés dans des services différents [14]. Ils avaient en revanche bénéficié d’une imagerie avec injection d’un produit de contraste dans le même service de radiologie et, pour la très grande majorité d’entre eux, par le même soignant. Cette technique permet donc de s’affranchir des écueils de l’enquête épidémiologique : mobilité des patients entre différents sites, exposition ponctuelle à des plateaux techniques, croisement fortuit entre deux patients… L’identification au préalable d’un lien génétique incite l’hygiéniste à s’acharner à trouver un lien entre les différents cas, alors qu’il n’aurait pas forcément insisté si les premiers éléments de l’enquête n’avaient montré aucun lien dans le temps et dans l’espace. Le Tableau I rapporte un certain nombre d’épidémies identifiées par l’approche sequence first [14,15,16,17].
La prochaine et peut-être ultime étape de ce changement de paradigme est l’association entre le séquençage sans a priori et l’intelligence artificielle : cette dernière permet en effet de traiter sans a priori les quantités extrêmement importantes de données démographiques et de laboratoire que chaque prise en charge hospitalière génère sous différentes formes (actes codifiés, comptes rendus de laboratoire, d’examen, de consultation ou d’hospitalisation…). Une étude récente a montré que cette association était bien plus performante et économique que le travail « à l’ancienne » d’un hygiéniste [15].
Conclusion
La comparaison des souches microbiennes reste un outil très utile dans le travail au quotidien de l’hygiéniste. En raison de la forte baisse de son coût, de la facilité de son interprétation et de l’utilisation de moyens humains, matériels et techniques partagés avec d’autres secteurs de l’hôpital, le séquençage de nouvelle génération constitue la référence indiscutable pour cette activité, au moins pour les bactéries. Demain, associée aux outils d’intelligence artificielle, sa déclinaison sans a priori constituera pour l’hygiéniste le point de départ de ses enquêtes d’évaluation de pratique et de son expertise sur la transmission croisée. Le séquençage de nouvelle génération sera alors à l’origine d’un véritable changement de paradigme et d’un tournant historique pour l’épidémiologie microbiologique hospitalière.
Notes :
1- Severe acute respiratory syndrome coronavirus 2, coronavirus 2 du syndrome respiratoire aigu sévère.
2- Acide désoxyribonucléique.
3- Coronavirus disease 2019, maladie à coronavirus 2019.
4- Repetitive sequence-based PCR, PCR basée sur des séquences répétitives.
5- Typage séquentiel multisite.
6- https://pubmlst.org/multilocus-sequence-typing (Consulté le 22-11-2022).
7- Spectrométrie de masse à temps de vol pour la désorption-ionisation laser assistée par matrice.
8- Polymorphisme d’un seul nucléotide.
9- Oxacillinase 48.
10- Centers for Disease Control and Prevention, centres pour le contrôle et la prévention des maladies (les CDC forment ensemble la principale agence fédérale des États-Unis en matière de protection de la santé publique – Wikipédia).
11- https://www.cdc.gov/pulsenet/index.html (Consulté le 22-11-2022).